 2019, Vol. 46
2019, Vol. 46扩展功能
文章信息
- 刘海燕, 王敬敬, 赵维, 李晴晴, 陈云, 易正炳, 吕新, 黄志勇
- LIU Hai-Yan, WANG Jing-Jing, ZHAO Wei, LI Qing-Qing, CHEN Yun, YI Zheng-Bing, LYU Xin, HUANG Zhi-Yong
- 塔里木河中下游流域棉田及胡杨林土壤细菌群落结构及多样性研究
- tructure of soil bacteria community and diversity in cotton field and Euphrates poplar forest in the middle and lower reaches of Tarim river basin
- 微生物学通报, 2019, 46(9): 2214-2230
- Microbiology China, 2019, 46(9): 2214-2230
- DOI: 10.13344/j.microbiol.china.180498
-
文章历史
- 收稿日期: 2018-06-26
- 接受日期: 2019-04-10
- 网络首发日期: 2019-04-22





2. 天津市工业生物系统与过程工程重点实验室 中国科学院天津工业生物技术研究所 天津 300308;
3. 新疆农垦科学院 新疆 石河子 832003;
4. 兵团第六师农业技术推广站 新疆 五家渠 831300
2. Tianjin Key Laboratory for Industrial Biological Systems and Bioprocessing Engineering, Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences, Tianjin 300308, China;
3. Xinjiang Agricultural Reclamation Academy of Sciences, Shihezi, Xinjiang 832003, China;
4. Agricultural Technology Promotion Station of the Sixth Division of the Xinjiang Production and Construction Corps, Wujiaqu, Xinjiang 831300, China
中国新疆的荒漠绿洲是山盆系统中最为精华的部分,占土地面积不到5%的绿洲聚集着新疆95%的人口与90%的社会财富[1-2]。20世纪50年代,我国在新疆地区进行大规模的农垦开发,在开垦之初大多以毁林毁草为代价,使得大面积的草地、荒漠植被和湿地等天然绿洲变成了人工绿洲,其中塔里木河流域就存在一定比例由胡杨林地转化而来的耕地[3]。大规模农垦活动造成了一系列生态问题,如自然植被退化、生物多样性降低、土壤次生盐渍化等。因此,关注和研究荒漠绿洲农业土壤的演变机制对于科学管理绿洲生态系统、维护人工绿洲土壤生态系统的稳定和可持续发展具有重要意义。
微生物作为一类多样性最高的生命形式,在地球化学循环(碳循环、氮循环、硫循环、磷循环和金属循环等)中承担着重要的生态功能。微生物复杂的群落结构、功能、相互作用及其动态变化对环境生态功能的维持有重要意义,土壤微生物群落的结构组成和特性在一定程度上反映了土壤生态系统的环境状况[4]。目前,国内外开展了较多关于荒漠[5-6]、盐碱地[7-8]、绿洲棉田[9-10]、森林[11-12]土壤的细菌多样性及分布特征的研究,然而,对单一生态系统细菌群落结构多样性的研究较多,对于不同生态系统间的对比研究则报道较少。在西北地区,李彦林等[13]对比研究了荒漠草原与耕地土壤可培养细菌多样性的差异,发现耕地土壤微生物多样性明显高于荒漠草原;李晨华等[14]研究了准葛尔盆地南缘盐生荒漠与绿洲农田土壤微生物群落组成变化特征,发现荒漠开垦为绿洲后,土壤细菌明显增加,放线菌显著减少,细菌在绿洲农田土壤矿化作用中占主导作用。从1990−2010年间,塔里木河中游人工绿洲、耕地增加了1 076.52%,主要由高覆盖草地、疏林地、低覆盖草地、中覆盖草地、有林地、灌木林地转化而来,天然绿洲所占比例越来越小[15]。然而,天然绿洲转换成人工绿洲后造成的生态影响,尤其是对土壤微生态的影响一直鲜有报道。本文选取塔里木河中下游流域由胡杨林地转化而来的耕地为研究对象,试图从土壤细菌群落多样性、群落结构变化、群落功能变化的角度,探究农垦开发对新疆绿洲土壤生态环境的影响。
1 材料与方法 1.1 材料 1.1.1 研究区域概况研究区位于新疆阿克苏地区塔里木河中下游流域,取样点在新疆生产建设兵团第一师阿拉尔垦区的绿洲棉田及附近的天然胡杨林中(40.50ʹ−40.83ʹN,81.15ʹ−81.48ʹE)。该区域积温4 113 ℃ (≥10 ℃),无霜期220 d,年平均气温7.2 ℃,年平均降水量170 mm,田间持水量30.1 g/m3,土壤容重1.2 g/cm3,供试土壤类型为灰漠土[16],质地为中壤。耕层土壤为20年以上的连作土壤,一般为40 cm,土壤养分基本情况:pH约为8.35,有机质19.09 g/kg,总盐3.75 g/kg,碱解氮51.5 mg/kg,有效磷19.89 mg/kg,速效钾212.8 mg/kg。
1.1.2 样品采集信息于2017年4月在春播前采集土壤样品,采样法为五点采样法:去除地表土,用直径5 cm的不锈钢土钻取0−20 cm的土壤样品,按“S”形取5钻,每一个点取样量大体一致,并均匀混合,保存于灭菌密封袋中。每个样品分别装到2个灭菌塑料袋中,一袋置于冰袋中运回实验室,储存在4 ℃中用于提取土壤总DNA;另一袋土样去除植物根系、石块等杂质,经自然风干、研磨过筛后储存在−20 ℃中以供土壤理化性质测定[17]。棉田(MT)的土壤样品采样点为ys-33 (40.66ʹN,81.31ʹE)、ys-36 (40.78ʹN,81.41ʹE)、ys-55 (40.73ʹN,81.21ʹE),胡杨林(HY)的土壤样品采样点为ys.8.1 (40.69ʹN,81.36ʹE)、ys.8.2 (40.75ʹN,81.40ʹE)、ys.8.3 (40.83ʹN,81.36ʹE)。
1.1.3 主要试剂和仪器细菌V4区16S rRNA基因扩增引物由生工生物工程(上海)股份有限公司合成,样品DNA提取试剂盒,MO BIO公司。NanoDrop 2000C分光光度计,Thermo公司;电泳仪、PCR仪,Bio-Rad公司。扩增子测序委托北京诺禾致源生物信息科技有限公司在新一代Illumina HiSeq 2500工作平台进行。
1.2 方法 1.2.1 土壤理化性质的测定土壤理化性质测定包括土壤全氮、总盐、有机质、碱解氮、有效磷、速效钾、pH等7个指标的具体实验方法参照《土壤农化分析与环境监测》[18],对每个土样所测指标进行3次平行测定,结果取其平均值。
1.2.2 土壤总DNA的提取及16S rRNA基因的PCR扩增将土壤样品液氮研磨3次,之后采用DNA提取试剂盒进行土壤总DNA提取。采用普通DNA产物纯化试剂盒进行DNA的纯化,经NanoDrop 2000C分光光度计和琼脂糖凝胶电泳测定其浓度和纯度。利用16S rRNA基因引物F515 (5′-GTGC CAGCMGCCGCGGTAA-3′)和R806 (5′-GGACTA CHVGGGTWTCTAAT-3′)[19]进行目的片段扩增。将扩增后样品寄往北京诺禾致源生物信息科技有限公司委托其进行新一代Illumina HiSeq 2500测序。
1.2.3 测序及分析Pearson相关系数分析[20]:采用Pearson (Centered)相关系数Pc来衡量两组分析对象线性关系的强弱,其取值范围为[0, 1],数学表达式为:
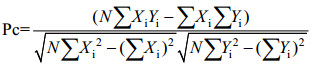
|
其中,X和Y分别对应两组分析对象的数据序列,N为样本个数,Pc绝对值越大,相关性越强。相关系数的绝对值越大,相关性越强:相关系数越接近于1或−1,相关度越强,相关系数越接近于0,相关度越弱。通常情况下通过以下取值范围判断变量的相关强度:相关系数0.8−1极强相关,0.6−0.8强相关,0.4−0.6中等程度相关,0.2−0.4弱相关,0.0−0.2极弱相关或无相关。
测序产生的双端序列使用FLASH软件进行拼接,参数默认[21]。拼接后序列使用QIIME软件进行数据过滤[22],过滤后序列利用USEARCH检测嵌合体,并使用filter_fasta.py命令将嵌合体去除[23]。利用UCLUST[24]对优质序列按相似度0.97进行聚类,并将其分配给可操作分类单元OTU (Operational taxonomic unit),选取每个OTU中丰度最高的序列为代表序列。OTU注释通过在QIIME中采用BLAST的方法与Greengenes数据库进行比对,获得每个OTU分类学信息。清除被分类到叶绿体、线粒体、真核生物或未知分类的序列以及单序列。为了保持数据的一致性,每个样本随机选择30 000条序列子集来计算样本之间的多样性和群落距离。
根据OTU列表中的各样品物种丰度情况,应用QIIME软件中的alpha_rarefaction.py命令[22],计算6种常用的生物多样性指数,样品的谱系多样性(Phylogenetic diversity,PD)、检测到的物种数(Observed species)、菌种丰富度指数(Chao1)、香农威纳指数(Shannon)、辛普森指数(Simpson)、覆盖率(Good’s coverage)等多样性评价指标。PD指某个地点所有物种间最短进化分支长度之和占各节点分支长度之和的比例;Observed species是指物种数目;Chao1为菌种丰富度指数,公式是:Chao1=Sobs+n1(n1–1)/2(n2+1),其中Chao1为估计的OTU数,Sobs为观测到的OTU数,n1为只有一条序列的OTU数目,n2为只有两条序列的OTU数目;Shannon为菌群多样性指数,公式是:H=–∑PilnPi,其中Pi为样品中属于第i种的个体的比例,如样品总个体数为N,第i种个体数为Ni,则Pi=Ni/N;Simpson为菌群多样性指数,公式是:D=1−∑(Ni/N)2,该指数描述从一个群落中连续两次抽样所得到的个体数属于同一种的概率,Ni为第i个物种的个体数,N为所有物种的个体数;Good’s coverage为测序深度指数,测序深度:C=1−n1/N,n1为只有含一条序列的OTU数目,N为抽样中出现的总序列数目。
根据OTU列表中的各样品物种丰度情况,应用QIIME软件中的summarize_taxa.py命令计算各样品在门、纲、目、科、属、种水平的物种组成。基于OTU表格进行主坐标分析(Principal coordinate analysis,PCoA),本文分别采用Jaccard (不考虑物种丰度)距离和Bray-Curtis (考虑物种丰度)距离计算各样品间的群落距离。基于线性判别分析流程[Linear discriminant analysis (LDA) effect size pipeline,LEfSe]确定每组的显著差异分类单元[25],该流程可在网址http://huttenhower.sph.harvard.edu/galaxy/获取,是一种用于发现高维生物标识的分析工具,用于区别两个或两个以上生物条件(或者是类群),它强调统计意义和生物相关性,能够在组与组之间寻找具有统计学差异的生物标识。该方法首先使用非参数因子克鲁斯卡尔-沃利斯秩和验检[Non-parametric factorial Kruskal-Wallis (KW) sum-rank test]检测不同分组间丰度差异显著的物种,再利用Wilcoxon秩和检验对每一组中的亚组进行两两检验,最后用线性判别分析(LDA)对数据进行降维并评估差异显著的物种的影响力(即LDA score),即用LDA值来估算每个组分(物种)丰度对差异效果影响的大小。利用FAPROTAX软件进行群落生态功能预测[26],该方法基于已发表验证的可培养微生物文献,因此预测准确度较好。
制图应用软件R、HEML、Past及Adobe illustrator。统计分析采用Excel、SPSS 19.0、R等。
2 结果与分析 2.1 土壤基本理化性质新疆绿洲棉田(MT)与原始胡杨林(HY)的土壤理化性质差异较大。绿洲棉田(MT)的土壤pH、总盐、全氮、碱解氮显著高于原始胡杨林(HY) (P < 0.05) (表 1)。绿洲棉田(MT)的土壤总盐、全氮、碱解氮含量约为原始胡杨林(HY)土壤的2−3倍,原始胡杨林(HY)全氮含量0.07±0.18 g/kg接近于新疆土壤全氮含量的最低值。
| Items | MT | HY |
| pH | 8.76±0.09a | 8.02±0.11b |
| 总盐Salinity (g/kg) | 4.25±0.52a | 1.84±0.29b |
| 有机质Organic matter (g/kg) | 8.37±2.62a | 8.05±0.13a |
| 全氮Total nitrogen (g/kg) | 0.20±0.11a | 0.07±0.18b |
| 碱解氮Alkaline hydrolysis nitrogen (mg/kg) | 91.58±5.16a | 27.53±7.26b |
| 有效磷Available phosphorous (mg/kg) | 37.69±4.76a | 24.70±10.16a |
| 有效钾Available potassium (mg/kg) | 125.39±22.74a | 123.41±47.19a |
| 注:表中数据为平均值±标准差;同行不同小写字母表示不同处理之间的土壤理化性质依据Duncan检测在0.05水平存在显著差异(n=3).新疆绿洲棉田(MT)和原始胡杨林(HY). Note: Data in the table are the Means±SD; The different normal letters in the same row indicate significant difference among treatments at 0.05 level (n=3) according to the Duncan test. Xinjiang oasis cotton field (MT) and original Euphrates poplar forest (HY). | ||
Pearson相关分析结果显示绿洲棉田(MT)的土壤总盐、pH与研究区域的土壤基本理化性质相比较相对较高,土壤盐含量与全氮、碱解氮和pH显著正相关(P < 0.05),土壤pH与全氮、碱解氮、有效磷、土壤盐分显著正相关(P < 0.05) (表 2)。
| Item | pH | Salt | OM | TN | AN | AP | AK |
| pH | 1 | ||||||
| Salt | 0.947* | 1 | |||||
| OM | 0.228 | −0.050 | 1 | ||||
| TN | 0.969** | 0.915* | 0.004 | 1 | |||
| AN | 0.971** | 0.924* | 0.010 | 1.000** | 1 | ||
| AP | 0.829* | 0.544 | 0.372 | 0.756 | 0.745 | 1 | |
| AK | 0.200 | 0.117 | 0.000 | 0.158 | 0.134 | 0.562 | 1 |
| 注:*: P < 0.05; **: P < 0.01. Salt:总盐含量;OM:有机质;TN:全氮含量;AN:碱解氮含量;AP:有效磷含量;AK:有效钾含量;新疆绿洲棉田(MT)和原始胡杨林(HY). Note: *: P < 0.05; **: P < 0.01. Salt: Total salt content; OM: Organic matter; TN: Total nitrogen; AN: Alkaline hydrolysis nitrogen; AP: Available phosphorous; AK: Available potassium. Xinjiang oasis cotton field (MT) and original Euphrates poplar forest (HY). | |||||||
微生物多样性分析的结果如图 1所示,6个样品的稀释曲线均基本趋于平缓,说明测序趋于饱和,取样合理,可真实反映出6个土壤样品中的微生物群落,结合各样品的覆盖率,说明测序结果包含了大多数微生物类群,基本能反映该区域土壤中微生物群落的结构组成,但仍有少量微生物种类未被发现。

|
| 图 1 3%水平样品的稀释曲线 Figure 1 Rarefaction curves of soil samples at cutoff level of 3% |
|
|
微生物的α多样性分析结果显示,新疆原始胡杨林(HY)土壤中的微生物多样性指数(Chao1、PD和Observed species)均显著高于绿洲棉田(MT) (P < 0.05) (表 3)。新疆原始胡杨林(HY)土壤中的微生物谱系多样性(PD)和观测到的OTU数目(Observed species)约为绿洲棉田(MT)的1.5倍,丰富度指数(Chao1)为绿洲棉田(MT)的2倍多。Shannon指数值越大,说明群落多样性越高;Simpson指数越大,说明群落多样性越低。虽然Shannon指数差异并不显著,但在胡杨林样品中仍高于棉田样品(表 4)。同时,各样品的Simpson指数均接近1,说明无论是胡杨林土壤样品还是棉田土壤样品中物种的多样性都相对较低,但是统计数据显示胡杨林样品的Simpson指数仍低于棉田样品(表 4)。
| 来源 Source |
编号 No. |
物种丰富度 Chao1 |
谱系多样性 PD |
OTU数目 Observed species |
香农指数 Shannon |
辛普森指数 Simpson |
覆盖率 Coverage (%) |
| MT | ys-33 | 9 266.13 | 451.97 | 4 506.90 | 10.09 | 1.00 | 91.90 |
| ys-36 | 9 463.16 | 485.31 | 4 770.40 | 10.08 | 1.00 | 91.68 | |
| ys-55 | 9 287.93 | 470.04 | 4 576.40 | 10.08 | 1.00 | 91.87 | |
| HY | ys.8.1 | 21 507.77 | 628.81 | 6 861.00 | 10.81 | 1.00 | 84.20 |
| ys.8.2 | 28 237.40 | 718.08 | 7 968.60 | 10.97 | 1.00 | 81.09 | |
| ys.8.3 | 15 875.36 | 536.51 | 5 514.40 | 9.41 | 0.97 | 87.82 | |
| 注:新疆绿洲棉田(MT)和原始胡杨林(HY). Note: Xinjiang oasis cotton field (MT) and original Euphrates poplar forest (HY). | |||||||
| Items | MT | HY |
| 丰富度指数Chao1 | 9 391±173b | 21 429±6 564a |
| 谱系多样性PD | 469±17b | 628±91a |
| OTU数目Observed species | 4 618±163b | 6 781±1 229a |
| 香农指数Shannon | 10.09±0.01a | 10.4±0.86a |
| 辛普森指数Simpson | 1±0a | 0.99±0.01a |
| 覆盖率Coverage (%) | 91.81±0b | 84.37±3a |
| 注:表中数据为平均值±标准差;同行不同小写字母表示不同处理之间的土壤细菌α多样性依据Duncan检测在0.05水平存在显著差异(n=3);新疆绿洲棉田(MT)和原始胡杨林(HY). Note: Data in the table are the Means±SD; The different normal letters in the same row indicate significant difference among treatments at 0.05 level (n=3) according to the Duncan test; Xinjiang oasis cotton field (MT) and original Euphrates poplar forest (HY). | ||
利用Illumina HiSeq 2500测序平台对6个土壤样品的微生物多样性进行检测,总共获得386 174个序列,每个样本平均45 000条序列(49 908−70 900)。将每个样本序列随机采取30 000条序列进行标准化分析后,发现优势物种为α-变形杆菌纲、放线菌门、芽单胞菌门、拟杆菌门、酸杆菌门、γ-变形菌纲、β-变形菌纲、δ-变形菌纲、绿弯菌门、厚壁菌门、浮霉菌门、泉古菌门和硝化螺旋菌门(丰度 > 1%) (图 2)。新疆绿洲棉田(MT)土壤中的酸杆菌门显著高于原始胡杨林(HY),δ-变形菌纲显著低于原始胡杨林(HY) (P < 0.05) (表 5)。新疆绿洲棉田(MT)土壤中酸杆菌门的含量约为原始胡杨林(HY)的2倍。新疆原始胡杨林(HY)土壤中δ-变形菌纲的含量约为绿洲棉田(MT)的2倍。

|
| 图 2 土壤微生物门水平(变形菌门按照纲分类)物种组成 Figure 2 Bacterial community composition on phylum level (phylum Proteobacteria is grouped into class level) 注:新疆绿洲棉田(MT)和原始胡杨林(HY). Note: Xinjiang oasis cotton field (MT) and original Euphrates poplar forest (HY). |
|
|
| Items | MT (%) | HY (%) |
| Alphaproteobacteria | 14.48±0.68a | 14.85±0.77a |
| Actinobacteria | 13.21±4.57a | 5.41±2.22a |
| Gemmatimonadetes | 9.95±1.76a | 7.83±1.56a |
| Bacteroidetes | 7.07±4.12a | 5.36±1.69a |
| Acidobacteria | 10.67±2.18a | 5.44±0.65b |
| Gammaproteobacteria | 12.53±4.94a | 21.27±9.61a |
| Betaproteobacteria | 6.49±1.81a | 8.08±1.05a |
| Deltaproteobacteria | 5.96±1.17b | 11.26±0.88a |
| Chloroflexi | 6.07±1.19a | 4.50±1.09a |
| Firmicutes | 4.20±1.99a | 2.26±0.59a |
| Planctomycetes | 3.26±1.81a | 3.11±0.72a |
| Crenarchaeota | 0.88±0.44a | 1.55±1.10a |
| Nitrospirae | 1.19±0.30a | 0.83±0.34a |
| Others | 4.05±0.47a | 8.26±1.87a |
| 注:表中数据为平均值±标准差;同行不同小写字母表示不同处理之间的土壤理化性质依据Duncan检测,在0.05水平存在显著差异(n=3);新疆绿洲棉田(MT)和原始胡杨林(HY). Note: Data in the table are the Means±SD; The different normal letters in the same row indicate significant difference among treatments at 0.05 level (n=3) according to the Duncan test. Xinjiang oasis cotton field (MT) and original Euphrates poplar forest (HY). | ||
在LEfSe分析结果中,进化分支图(图 3A)中由内至外的圆圈代表了由门至属的分类级别,不同的分类级别上的每一个小圆圈代表该水平下的一个分类,小圆圈的直径大小代表了相对丰度的大小。无显著差异的物种统一着色为黄色,差异显著的物种Biomarker跟随组别进行着色,红色节点表示在原始胡杨林(HY)组别中起到重要作用的微生物类群,绿色节点表示在绿洲棉田(MT)组别中起到重要作用的类群。选取LDA score大于2.0的为显著差异物种,具有统计学差异的Biomaker共计301种,选择LDA score大于3.5的差异物种进行LDA分布柱状图展示(图 3B),其中柱状图的颜色代表各自的组别,长短代表的是LDA score,即不同组间显著差异物种的影响程度,例如放线菌门(Actinobacteria)、酸杆菌门(Acidobacteria)、鞘脂单胞菌目(Sphingomonadales)、脱硫杆菌目(Desulfobacterales)、地杆菌属(Geobacter)是导致绿洲棉田(MT)异于原始胡杨林(HY)的主要微生物物种。

|
| 图 3 土壤微生物LEfSe分析的进化分支图(A)和LDA值(B) (LDA值> 3.5) Figure 3 Least discriminant analysis (LDA) effect size taxonomic cladogram (A) and LDA score of the discriminants comparing soil bacteria 注:新疆绿洲棉田(MT)和原始胡杨林(HY). Note: Xinjiang oasis cotton field (MT) and original Euphrates poplar forest (HY). |
|
|
新疆绿洲棉田(MT)土壤中富集的微生物主要包括酸杆菌门、放线菌门、拟杆菌门、绿弯菌门、厚壁菌门、变形菌门以及泉古菌门中的一些物种(图 3),如酸杆菌门中的iii1_15;放线菌门中的微球菌科(Micrococcaceae)、类诺卡氏菌科(Nocardioidaceae)、MB_A2_108、Thermoleophilia、Gaiellales、Gaiellaceae、Solirubrobacterales;拟杆菌门中的黄杆菌纲(Flavobacteriia)、黄杆菌目(Flavobacteriales);厚壁菌门的Allobaculum、α-变形菌纲的鞘脂单胞菌目(Sphingomonadales)、Kaistobacter;γ-变形菌纲的黄色单胞菌目(Xanthomonadales)、溶杆菌属(Lysobacter);来自泉古菌门的Nitrososphaerales、亚硝化暖菌属(Candidatus Nitrososphaera)。
新疆原始胡杨林(HY)土壤中富集的微生物主要包括绿菌门、δ-变形菌纲和变形菌门中的其他一些物种,如属于δ-变形菌纲的脱硫杆菌目(Desulfobacterales)、脱硫杆菌科(Desulfobulbaceae)、除硫单胞菌目(Desulfuromonadales)、地杆菌科(Geobacteraceae)、地杆菌属(Geobacter)、粘土杆菌科(Pelobacteraceae);属于γ-变形菌的交替单胞菌目(Alteromonadales)、着色菌目(Chromatiales)、硫发菌目(Thiotrichales)、鱼立克次体科(Piscirickettsiaceae);属于β-变形菌的红环菌目(Rhodocyclales)、红环菌科(Rhodocyclaceae);来自奇古菌门(Thaumarchaeota)的餐古菌目(Cenarchaeales)、餐古菌科(Cenarchaeaceae)的亚硝化侏儒菌属(Nitrosopumilus)。
PCoA分析结果显示新疆绿洲棉田(MT)和原始胡杨林(HY)土壤中的微生物群落结构明显不同(图 4)。其中,无论是基于Jaccard距离的PCoA分析还是基于Bray-Curtis距离的PCoA分析均显示棉田样品(ys-33、ys-36、ys-55)能够较为紧密的聚集在一起,且明显与胡杨林样品分开;胡杨林样品中的ys.8.3与其他两个样品(ys.8.1和ys.8.2)距离较远。

|
| 图 4 基于Jaccard (A)和Bray-Curtis (B)距离算法的土壤微生物群落PCoA分析 Figure 4 Principle coordinate analysis based on Jaccard (A) and Bray-Curtis (B) distance method of soil bacteria community structure 注:新疆绿洲棉田(MT)和原始胡杨林(HY). Note: Xinjiang oasis cotton field (MT) and original Euphrates poplar forest (HY). |
|
|
利用FAPROTAX软件基于种分类水平的群落组成进行生态功能预测,结果显示1 469个种水平的细菌分类单元中有426条物种信息可以被预测,1 007条不可预测,可预测比例为31.45%。功能分析的结果显示,新疆绿洲棉田(MT)和原始胡杨林(HY)土壤中的微生物群落功能主要有化能自养、好氧化能自养、发酵、硝化、硝化还原、硝化呼吸、氮呼吸、光养、好氧亚硝酸盐氧化、光能自养、亚硝酸盐呼吸、无氧光自养硫氧化、无氧光自养、光能异养和好氧氨氧化(丰度 > 1%)。HeatMap分析结果显示新疆绿洲棉田(MT)和原始胡杨林(HY)土壤中的微生物群落功能明显不同。绿洲棉田(MT)的3个样品聚为一组,原始胡杨林(HY)三个样品聚为一组(图 5)。新疆绿洲棉田(MT)土壤中微生物群落的好氧化能自养、硝化、光能异养、好氧氨氧化、铁呼吸、尿素分解等功能显著高于原始胡杨林(HY),甲烷营养、铁氧化暗反应等功能显著低于原始胡杨林(HY) (P < 0.05) (表 6)。其中,根据功能预测结果文件显示,与硝化作用相关的功能主要来自泉古菌门的Candidatus Nitrososphaera、β-变形菌纲的亚硝化单胞菌目(Nitrosomonadales)以及硝化螺旋菌门(Nitrospirae),好氧氨氧化作用主要来自前三者。尿素分解作用主要来自α-变形菌的根瘤菌目(Rhizobiales)和γ-变形菌的黄色单胞菌目(Xanthomonadales)。绿洲棉田(MT)的铁呼吸、尿素分解等功能显著,说明秋季绿洲棉田(MT)栽培管理加强铃肥和根外追肥,达到稳产高产的同时土壤营养富集[27],春季绿洲棉田硝化作用高于原始胡杨林(HY),说明人为施肥提高了硝化细菌多样性,微生物功能差异最先起到指示作用。

|
| 图 5 土壤微生物群落功能HeatMap图 Figure 5 HeatMap of soil bacterial community function 注:新疆绿洲棉田(MT)和原始胡杨林(HY). Note: Xinjiang oasis cotton field (MT) and original Euphrates poplar forest (HY). |
|
|
| Community function | MT (%) | HY (%) |
| Chemoheterotrophy | 16.83±5.75a | 8.82±1.06a |
| Aerobic_chemoheterotrophy | 13.16±2.61a | 7.73±1.07b |
| Phototrophy | 1.13±0.05a | 1.10±0.25a |
| Photoautotrophy | 0.96±0.10a | 0.98±0.31a |
| Photoheterotrophy | 0.91±0.09a | 0.70±0.05b |
| Anoxygenic_photoautotrophy_S_oxidizing | 0.81±0.18a | 0.97±0.31a |
| Iron_respiration | 0.09±0.05a | 0.89±0.36b |
| Respiration_of_sulfur_compounds | 0.12±0.04a | 0.82±0.54a |
| Sulfate_respiration | 0.11±0.05a | 0.81±0.55a |
| Dark_hydrogen_oxidation | 0.30±0.17a | 0.37±0.07a |
| Dark_oxidation_of_sulfur_compounds | 0.01±0.00a | 0.44±0.30a |
| Dark_sulfide_oxidation | 0.00±0.00a | 0.44±0.30a |
| Dark_iron_oxidation | 0.00±0.00b | 0.15±0.06a |
| Fermentation | 2.95±2.22a | 0.73±0.11a |
| Nitrification | 2.45±0.78a | 1.09±0.28b |
| Nitrate_reduction | 1.31±0.03a | 2.06±0.40a |
| Nitrogen_respiration | 1.06±0.09a | 1.71±0.56a |
| Aerobic_nitrite_oxidation | 1.20±0.30a | 0.84±0.34a |
| Nitrite_respiration | 0.78±0.19a | 1.07±0.13a |
| Aerobic_ammonia_oxidation | 1.25±0.61a | 0.25±0.07b |
| Nitrous_oxide_denitrification | 0.77±0.20a | 0.59±0.04a |
| Nitrogen_fixation | 0.05±0.04a | 0.02±0.01a |
| Chitinolysis | 1.16±1.22a | 0.20±0.09a |
| Aromatic_compound_degradation | 0.47±0.47a | 0.18±0.04a |
| Ureolysis | 0.38±0.08a | 0.16±0.08b |
| Methanotrophy | 0.06±0.01b | 0.11±0.01a |
| 注:表中数据为平均值±标准差;同行不同小写字母表示不同处理之间的土壤细菌群落功能依据Duncan检测在0.05水平存在显著差异(n=3);新疆绿洲棉田(MT)和原始胡杨林(HY). Note: Data in the table are the Means±SD; The different normal letters in the same row indicate significant difference among treatments at 0.05 level (n=3) according to the Duncan test; Xinjiang oasis cotton field (MT) and original Euphrates poplar forest (HY). | ||
根据前向选择和统计分析的结果,选择全氮、盐分、pH、有机质和微生物物种(门水平,变形菌纲水平)进行RDA分析(图 6)。结果显示全氮对群落变异的解释度为55.7% (P=0.004),盐分为21.5% (P=0.146),有机质为16.9% (P=0.078),pH为3.9% (P=0.294)。因此可以推测全氮是驱动群落变异的主要因子。全氮、盐分、pH与放线菌门、酸杆菌门、绿弯菌门、浮霉菌门、厚壁菌门、芽单胞菌门正相关。有机质与变形菌门、泉古菌门、拟杆菌门等正相关。

|
| 图 6 土壤微生物和理化特性的RDA分析 Figure 6 Redundancy analysis of soil bacterial community and soil physical and chemical properties 注:新疆绿洲棉田(MT)和原始胡杨林(HY). Note: Xinjiang oasis cotton field (MT) and original Euphrates poplar forest (HY). |
|
|
棉田长期过量施用化肥,尤其是氮、磷、钾肥,会造成严重的土壤盐碱化,并导致微生物多样性显著降低,使得放线菌、酸杆菌、绿弯菌、浮霉菌、厚壁菌、芽单胞菌等参与氮循环、光合作用的微生物明显增加,参与甲烷循环、铁循环的变形菌、泉古菌、拟杆菌等微生物明显减少。
3 讨论新疆棉田一直是农业微生物研究者关注的对象,以往对新疆棉田的细菌学研究主要集中在棉田本身的微生物特性研究上[10, 28-30],而少有报道针对棉田生态系统与其他自然生态系统的对比研究。自从20世纪50年代,我国在新疆进行大规模农垦开发以来,对当地绿洲生态系统产生了一系列影响。本文通过对塔里木河中下游流域绿洲棉田与原始胡杨林土壤中的细菌群落结构进行比较研究,发现该地区土壤细菌群落在人类长期农业发展中具有显著变化,长期耕种施肥等人为因素干扰会导致细菌群落发生改变。
本研究的土壤pH在7.92−8.79之间,与郑贺云等[31]等对新疆阿克苏地区盐碱地土壤pH (7.57−8.75,碱性水平)的报道较为一致;另外,本研究中的胡杨林土壤的总盐、有机质、速效钾含量与王新英等[32]对塔里木河流域胡杨林土壤理化性质的研究结果有较高的一致性;同时,本研究中的棉田土壤总盐、有机质和全氮含量与顾美英等[10]对新疆绿洲农田不同连作年限棉花根际土壤的结果也较为接近。综上所述,本研究中涉及的新疆棉田土壤样品和胡杨林样品具有一定的代表性,样品中的微生物多样性及细菌群落结构差异在一定程度上能够反映两种不同生态系统的微生物群落变化。
微生物多样性指数可以反映群落多样性的变化,Observed species反映的是样品中可检测到的OTU数;Chao1指数用来反映物种丰富度,对稀有物种很敏感(Singletons和Doubletons);PD则反映的是谱系多样性,即系统发育多样性。Shannon指数和Simpson指数考察群落物种多样性。本文使用这几个指标衡量棉田生态系统和胡杨林生态系统的细菌多样性差异,既考虑物种的丰富度、均匀度,也考虑了系统进化差异,前三类指数在棉田生态系统中均显著低于胡杨林生态系统,说明长期的耕作种植行为使棉田土壤中的微生物种类趋于简化,即物种丰富度降低、系统发育多样性也降低。同时,Shannon指数也显示了同样的变化规律,这与顾美英等[10]对新疆绿洲农田不同连作年限棉花根际土壤微生物群落多样性的研究结果较为一致,新开垦棉田(连作1年)的土壤微生物Shannon指数最高,随着连作年限的延长,连作5年略有下降,随后上升,连作10年以后逐年下降,即随着人为影响时间的延长,微生物群落的Shannon指数逐渐降低。但是同类型的研究尚未观测物种多度指数(Chao1)、物种数(Observed species)以及系统发育指数(PD),在本研究中,这3个指数在长期耕作的棉田系统中显著低于原始胡杨林系统,尤其Chao1指数相差一倍以上,说明长期人为干扰对生态多样性的影响在物种种类变化上尤为显著。
关于长期耕作导致的棉田土壤微生物群落结构变化的研究,主要有Zhang等[33]从南疆阿瓦提土壤细菌生态学的角度分析了作物种植对土壤细菌群落的影响,结果显示随着人为耕作时间的延长,棉花根区微生物群落中酸杆菌门(Acidobacteria)、硝化螺旋菌门(Nitrospirae)、泉古菌门(Crenarchaeota)和疣微菌门(Verrucomicrobia)显著增高,α-变形菌纲(Alphaproteobacteria)、芽单胞菌门(Gemmatimonadetes)、奇古菌门(Thaumarchaeota)、Gemm-1、δ-变形菌纲(Deltaproteobacteria)以及Acidobacteria-6均明显升高,在目水平黄色单胞菌目(Xanthomonadales)、鞘脂单胞菌目(Sphingomonadales)以及Nitrososphaerales显著增高。在本文中,棉田样品相对于原始胡杨林样品显著增高的有酸杆菌门、属于α-变形菌的鞘脂单胞菌目(Sphingomonadales)、属于γ-变形菌的黄色单胞菌目(Xanthomonadales)、属于泉古菌门的Nitrososphaerales,与上述报道结果是一致的。说明该研究中棉田相对于胡杨林的土壤细菌群落变化很大程度上是由长期耕作导致的。关于新疆胡杨林微生物多样性的报道主要集中在对可培养细菌及胡杨内生细菌的分离研究中[34-35],使用高通量测序技术对胡杨林土壤细菌群落结构进行的研究较少。吾尔麦提汗·麦麦提明等[34]对塔里木胡杨林可培养胡杨内生细菌多样性的研究结果显示,在分离的588株胡杨内生细菌中γ-变形菌纲所占比例最高达50%以上,其次是厚壁菌门(> 30%)、放线菌门(> 10%)、α-变形菌纲(4%)、拟杆菌门(0.5%)、β-变形菌纲(0.34%),γ-变形菌纲和厚壁菌门的细菌是胡杨茎秆液中的优势类群,假单胞菌属(29.76%)和芽孢杆菌属(19.05%)为优势属。吐尔逊阿依·麦麦提依明等[35]在对塔里木胡杨林土壤中可培养细菌多样性的研究结果显示,在分离得到的193株土壤细菌中厚壁菌门占总分离菌的比例最高(> 50%),其次是γ-变形菌(约30%)、放线菌门(7.3%)、β-变形菌纲(2.6%)、α-变形菌纲(1.6%)。在本研究的结果中,胡杨林土壤样品共测序得到11 490个OTU,经物种分类后显示比例最高的是γ-变形菌纲(> 20%),其次是α-变形菌纲(> 10%)、δ-变形菌纲(> 10%)、β-变形菌纲(8%)、芽单胞菌门(7%)、酸杆菌门(5%)、放线菌门(5%)、拟杆菌门(5%)、绿弯菌门(4%)、浮霉菌门(3%)、厚壁菌门(2%)、泉古菌门(1.5%)。相较于他人的研究,使用高通量测序技术获得了更多的胡杨林土壤细菌物种分类信息,虽然各大类比例不尽相同,但是主要的优势类群是一致的,除厚壁菌门相对比例较低,这可能与上述报道中分离方法的偏倚性有关。此外,功能预测的结果显示胡杨林样品中参与甲烷循环、铁循环的功能比棉田中显著富集,说明在胡杨林系统中较好地保持了原始生态环境中的微生物种类和功能。
棉田样品和胡杨林样品群落结构的对比显示,两种生态系统在门水平上的优势类群中主要存在显著的比例差异,同时主坐标分析的结果不仅显示出两类样品在群落结构上的差异,同时还显示出取自棉田样品群落的一致性较高,而取自胡杨林的土壤样品群落差异较大。这与Rodrigues等[36]研究结果相一致,即森林向农业转化后,土壤细菌的β多样性降低,群落结构趋于一致。这说明,该研究中的土壤微生物群落分别在α多样性、群落组成、群落结构差异(β多样性)方面显示出由原始胡杨林环境向长期耕作的人工绿洲环境转变后的响应。
该研究试图利用取样点的土壤理化性质来寻找两类生态系统对群落结构产生影响的主要因子。RDA分析结果显示,土壤全氮对群落变异的解释度最高也最显著,且全氮含量主要对棉田系统细菌群落中的酸杆菌门、绿弯菌门、硝化螺旋菌门、浮霉菌门、拟杆菌门、放线菌门有正向影响。同时,功能预测的结果,在棉田样品中显著富集的Candidatus Nitrososphaera、亚硝化单胞菌目(Nitrosomonadales)均对硝化作用和好氧氨氧化作用有贡献,而α-变形菌的根瘤菌目(Rhizobiales)以及γ-变形菌的黄色单胞菌目(Xanthomonadales)则对尿素分解有影响。说明,土壤中的微生物群落结构会对环境中增加的氮进行反馈,通过调整相应功能的微生物类群来适应不断增加的外源营养物。Cheng等[37]对玛纳斯河流域弃耕地复耕后土壤理化性质对群落差异影响的研究显示,氨氮对酸杆菌门、硝化螺旋菌门具有正向影响,这个结果与本文的研究结果一致。除了氮元素,盐分对群落差异也有较大影响,盐分的解释度排在第二位。研究区域本身降水稀少,日蒸发量大,造就的盐碱环境,盐度对群落的影响也是不容忽视的一部分。虽然盐度单独解释的部分并不显著,但由于土壤盐含量与全氮、碱解氮的高度线性关系,盐度对优势类群的影响与全氮是一致的,也对酸杆菌门、放线菌门有正向影响,其中,放线菌中含有大量耐盐菌。郑贺云等[31]对新疆阿克苏地区盐碱地细菌类群多样性研究结果表明,在分离到的嗜盐菌中存在较高比例的放线菌,关统伟等[8]、李文均等[38]、Xu等[39]也均有相关报道。在本研究中,棉田土壤中盐度显著高于胡杨林,其放线菌在门水平上也显著高于胡杨林土壤样品;然而,群落功能预测的结果显示,参与氮代谢的微生物并不包含放线菌。因此,RDA结果表明土壤盐分导致放线菌与全氮、盐度和pH的高度正相关性。
总之,本研究从细菌群落结构变化的角度探讨了棉田和胡杨林土壤样品的差异,实际上取样的棉田在进行耕作以前也是胡杨林的一部分,在毁林造田的巨大人为扰动下造就了现今的棉田生态系统和胡杨林生态系统。在一定程度上,本文研究的棉田样品可以代表当地的农业生态系统,也即是本文实际上在探究农业活动对土壤微生物多样性的影响和变化规律。尽管在样品数量的选择上有一定的局限性,但是每个样品均是通过五点采样、进行3次重复后混匀获得的土样,这保证了实验数据的严谨性。此外,除了分析群落结构的差异,还初步进行了群落功能的预测,预测结果主要用来辅助解释导致群落差异的因素。后续将会进一步增加取样点和生物学重复,在定量功能基因的角度进行更多的研究,多方面阐述农业活动对生态系统的干扰。
4 结论通过对塔里木河中下游流域绿洲棉田及原始胡杨林土壤理化性质、α多样性、β多样性、细菌群落组成及其与环境因子关系的研究,发现:(1)新疆绿洲棉田与原始胡杨林的土壤理化性质差异较大:绿洲棉田的土壤pH、总盐、全氮、碱解氮显著高于原始胡杨林,其土壤总盐、全氮、碱解氮含量约为原始胡杨林土壤的2−3倍;(2)新疆绿洲棉田细菌群落α多样性显著低于原始胡杨林;(3)细菌群落中的酸杆菌门、放线菌门、黄杆菌纲、绿弯菌门、厚壁菌门和α-变形菌中的鞘脂单胞菌目、γ-变形菌中的黄色单胞菌目、溶杆菌属,以及来自泉古菌门的Nitrososphaerales、亚硝化暖菌属在棉田土壤中显著富集,而绿菌门、δ-变形菌中的脱硫杆菌目、除硫单胞菌目和γ-变形菌中的交替单胞菌目、着色菌目、硫发菌目、属于β-变形菌的红环菌目以及来自奇古菌门的亚硝化侏儒菌属在胡杨林土壤中显著富集;(4)棉田土壤和胡杨林土壤的细菌群落结构差异明显,且棉田土壤中的细菌群落结构趋于一致;(5)土壤全氮对两种生态系统土壤中的细菌群落结构差异的解释度最大。以上结果反映了在长期人为干扰下绿洲棉田系统相对于原始胡杨林系统土壤微生态改变的情况,通过对特定土壤微生物类群的追踪或可评估土壤生态系统受人为耕种、施肥等开发活动的影响程度,可为后续对新疆绿洲生态系统土壤评价提供参考。
致谢: 本试验得到兵团农业技术推广站各级工作人员的大力配合与支持,在此一并深表谢意!| [1] |
Wang JY, Wang ML, Zhang FH. Soil microbial properties under typical halophytic vegetation communities in arid regions[J]. Acta Ecologica Sinica, 2016, 36(8): 2363-2372. (in Chinese) 王静娅, 王明亮, 张凤华. 干旱区典型盐生植物群落下土壤微生物群落特征[J]. 生态学报, 2016, 36(8): 2363-2372. |
| [2] |
Zhao AD, Xu S, Zeng W, et al. Analysis of unstable farmland in arid and semi-arid regions and feasibility evaluation of its conversion[J]. Transactions of the Chinese Society of Agricultural Engineering, 2016, 32(17): 215-225. (in Chinese) 赵爱栋, 许实, 曾薇, 等. 干旱半干旱区不稳定耕地分析及退耕可行性评估[J]. 农业工程学报, 2016, 32(17): 215-225. DOI:10.11975/j.issn.1002-6819.2016.17.029 |
| [3] |
Gui DW, Lei JQ, Mu GJ, et al. Effects of different management intensities on soil quality of farmland during oasis development in southern Tarim Basin, Xinjiang, China[J]. International Journal of Sustainable Development & World Ecology, 2009, 16(4): 295-301. |
| [4] |
van Der Heijden MGA, Wagg C. Soil microbial diversity and agro-ecosystem functioning[J]. Plant and Soil, 2013, 363(1/2): 1-5. |
| [5] |
Neilson JW, Quade J, Ortiz M, et al. Life at the hyperarid margin: novel bacterial diversity in arid soils of the Atacama Desert, Chile[J]. Extremophiles, 2012, 16(3): 553-566. DOI:10.1007/s00792-012-0454-z |
| [6] |
Saul-Tcherkas V, Unc A, Steinberger Y. Soil microbial diversity in the vicinity of desert shrubs[J]. Microbial Ecology, 2013, 65(3): 689-699. DOI:10.1007/s00248-012-0141-8 |
| [7] |
Niu SQ, Long Y, Li HY, et al. Microbial diversity in saline alkali soil from Hexi Corridor analyzed by Illumina MiSeq high-throughput sequencing system[J]. Microbiology China, 2017, 44(9): 2067-2078. (in Chinese) 牛世全, 龙洋, 李海云, 等. 应用Illumina MiSeq高通量测序技术分析河西走廊地区盐碱土壤微生物多样性[J]. 微生物学通报, 2017, 44(9): 2067-2078. |
| [8] |
Guan TW, Wu JY, Tang SK, et al. Phylogenetic diversity of halophilic actinomycetes from hypersaline environments in Tarim Basin of Xinjiang, China[J]. Microbiology China, 2008, 35(11): 1698-1702. (in Chinese) 关统伟, 吴晋元, 唐蜀昆, 等. 新疆塔里木盆地可培养嗜盐放线菌系统发育多样性[J]. 微生物学通报, 2008, 35(11): 1698-1702. |
| [9] |
Zhang W, Chen YF. Analysis of the bacterial communities in continuous cotton fields of Xinjiang Province[J]. Acta Ecologica Sinica, 2014, 34(16): 4682-4689. (in Chinese) 张伟, 陈一峰. 棉花长期连作对新疆土壤细菌群落结构的影响[J]. 生态学报, 2014, 34(16): 4682-4689. |
| [10] |
Gu MY, Xu WL, Mao J, et al. Microbial community diversity of rhizosphere soil in continuous cotton cropping system in Xinjiang[J]. Acta Ecologica Sinica, 2012, 32(10): 3031-3040. (in Chinese) 顾美英, 徐万里, 茆军, 等. 新疆绿洲农田不同连作年限棉花根际土壤微生物群落多样性[J]. 生态学报, 2012, 32(10): 3031-3040. |
| [11] |
Xu MB. Study on diversity of culturable bacteria isolated from soil of rhizosphere of Populus euphratic forest and polyphasic taxonomy approach of two novel species[D]. Wuhan: Doctoral Dissertation of Wuhan University, 2011 (in Chinese) 许孟博.新疆胡杨林根际可培养细菌的多样性及两个潜在新分类单元的多相分类学研究[D].武汉: 武汉大学博士学位论文, 2011 |
| [12] |
Qiao HL, Tian CM, Luo YQ, et al. Diversity of soil microorganism in the natural Populus euphratica forests in Xinjiang, northwestern China[J]. Journal of Beijing Forestry University, 2007, 29(5): 127-131. (in Chinese) 乔海莉, 田呈明, 骆有庆, 等. 新疆天然胡杨林土壤微生物多样性的研究[J]. 北京林业大学学报, 2007, 29(5): 127-131. DOI:10.3321/j.issn:1000-1522.2007.05.025 |
| [13] |
Li YL, Chen JX, Zhou YT, et al. Diversity and distribution of culturable microbial communities of semiarid desert steppe and cultivated land in northwestern China[J]. Journal of Agricultural Resources and Environment, 2016, 33(3): 244-252. (in Chinese) 李彦林, 陈吉祥, 周永涛, 等. 西北半干旱荒漠草原与耕地土壤可培养微生物多样性及分布特征比较[J]. 农业资源与环境学报, 2016, 33(3): 244-252. |
| [14] |
Li CH, Li Y, Xie JX, et al. Comparative on microbial community composition and microbial activities in desert and oasis soils[J]. Acta Ecologica Sinica, 2007, 27(8): 3391-3399. (in Chinese) 李晨华, 李彦, 谢静霞, 等. 荒漠-绿洲土壤微生物群落组成与其活性对比[J]. 生态学报, 2007, 27(8): 3391-3399. DOI:10.3321/j.issn:1000-0933.2007.08.036 |
| [15] |
Chen CQ, Wu Y, Wang J, et al. Artificial and natural oasis mutual converting process and the proper ration in the middle reaches of Tarim River basin[J]. Journal of Water Resources & Water Engineering, 2017, 28(3): 42-47, 54. (in Chinese) 陈超群, 吴煜, 王健, 等. 塔里木河中游人工与天然绿洲相互转化过程与适宜比例探讨[J]. 水资源与水工程学报, 2017, 28(3): 42-47, 54. |
| [16] |
Zhang H, Wu JW, Zheng QH, et al. A preliminary study of oasis evolution in the Tarim Basin, Xinjiang, China[J]. Journal of Arid Environments, 2003, 55(3): 545-553. DOI:10.1016/S0140-1963(02)00283-5 |
| [17] |
Zhang HY, He JZ, Xu B, et al. Variety of soil microbial structure in continuous cropping cotton field in South Xinjiang[J]. Microbiology China, 2010, 37(5): 689-695. (in Chinese) 张海燕, 贺江舟, 徐彪, 等. 新疆南疆不同连作年限棉田土壤微生物群落结构的变化[J]. 微生物学通报, 2010, 37(5): 689-695. |
| [18] |
Yang JH, Wang CL, Dai HL. Soil Agrochemical Analysis and Environmental Monitoring[M]. Beijing: China Land Press, 2008. (in Chinese) 杨剑虹, 王成林, 代亨林. 土壤农化分析与环境监测[M]. 北京: 中国大地出版社, 2008. |
| [19] |
Caporaso JG, Lauber CL, Walters WA, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample[J]. Proceedings of the National Academy of Sciences of the United States of America, 2011, 108(S1): 4516-4522. |
| [20] |
Jiang H, Deng WM, Chen XQ. Analysis of wine based on pearson coefficient and multiple kernel support vector classification[J]. Transactions of the Chinese Society for Agricultural Machinery, 2014, 45(1): 203-208. (in Chinese) 蒋辉, 邓伟民, 陈晓青. 基于Pearson系数与多元核支持向量分类的葡萄酒分析[J]. 农业机械学报, 2014, 45(1): 203-208. |
| [21] |
Magoč T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies[J]. Bioinformatics, 2011, 27(21): 2957-2963. DOI:10.1093/bioinformatics/btr507 |
| [22] |
Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data[J]. Nature Methods, 2010, 7(5): 335-336. DOI:10.1038/nmeth.f.303 |
| [23] |
Edgar RC, Haas BJ, Clemente JC, et al. UCHIME improves sensitivity and speed of chimera detection[J]. Bioinformatics, 2011, 27(16): 2194-2200. DOI:10.1093/bioinformatics/btr381 |
| [24] |
Edgar RC. Search and clustering orders of magnitude faster than BLAST[J]. Bioinformatics, 2010, 26(19): 2460-2461. DOI:10.1093/bioinformatics/btq461 |
| [25] |
Segata N, Izard J, Waldron L, et al. Metagenomic biomarker discovery and explanation[J]. Genome Biology, 2011, 12(6): R60. DOI:10.1186/gb-2011-12-6-r60 |
| [26] |
Louca S, Parfrey LW, Doebeli M. Decoupling function and taxonomy in the global ocean microbiome[J]. Science, 2016, 353(6305): 1272-1277. DOI:10.1126/science.aaf4507 |
| [27] |
Gu YF, Zhang XP, Tu SH, et al. Effect of long-term fertilization on nitrification and nitrobacteria community in a purple paddy soil under rice-wheat rotations[J]. Acta Ecologica Sinica, 2008, 28(5): 2123-2130. (in Chinese) 辜运富, 张小平, 涂仕华, 等. 长期定位施肥对紫色水稻土硝化作用及硝化细菌群落结构的影响[J]. 生态学报, 2008, 28(5): 2123-2130. DOI:10.3321/j.issn:1000-0933.2008.05.028 |
| [28] |
Gu MY, Liu HL, Li ZQ, et al. Impact of biochar application on soil nutrients and microbial diversities in continuous cultivated cotton fields in Xinjiang[J]. Scientia Agricultura Sinica, 2014, 47(20): 4128-4138. (in Chinese) 顾美英, 刘洪亮, 李志强, 等. 新疆连作棉田施用生物炭对土壤养分及微生物群落多样性的影响[J]. 中国农业科学, 2014, 47(20): 4128-4138. DOI:10.3864/j.issn.0578-1752.2014.20.021 |
| [29] |
Wang DM, Ding RF, Li HQ, et al. Changes in rhizosphere soil microbial community in Xinjiang Bt and non-Bt cotton fields[J]. Xinjiang Agricultural Sciences, 2014, 51(5): 885-892. (in Chinese) 王冬梅, 丁瑞丰, 李海强, 等. 新疆Bt棉田根际土壤微生物群落变化研究[J]. 新疆农业科学, 2014, 51(5): 885-892. |
| [30] |
Xu WX, Luo M, Li DP, et al. Changes in soil physicochemical properties and microflorae under long-term cotton continuous cropping[J]. Agricultural Research in the Arid Areas, 2014, 32(3): 134-138. (in Chinese) 徐文修, 罗明, 李大平, 等. 不同连作年限棉田土壤理化性质及微生物区系变化规律研究[J]. 干旱地区农业研究, 2014, 32(3): 134-138. |
| [31] |
Zheng HY, Li ZK, Li C, et al. Analysis of the bacterial diversity and dominant population in Akesu Saline-alkali in Xinjiang[J]. Microbiology China, 2012, 39(7): 1031-1043. (in Chinese) 郑贺云, 黎志坤, 李超, 等. 新疆阿克苏地区盐碱地细菌类群多样性及优势菌群分析[J]. 微生物学通报, 2012, 39(7): 1031-1043. |
| [32] |
Wang XY, Shi JH, Liu MX. Physicochemical properties and correlations of the soils in the Populus euphratica forests of different ages in the Tarim River Basin[J]. Journal of Northeast Forestry University, 2016, 44(9): 63-68. (in Chinese) 王新英, 史军辉, 刘茂秀. 塔里木河流域不同龄组胡杨林土壤理化性质及相关性[J]. 东北林业大学学报, 2016, 44(9): 63-68. DOI:10.3969/j.issn.1000-5382.2016.09.014 |
| [33] |
Zhang W, Du Y. Analysis of the succession of structure of the bacteria community in soil from long-term continuous cotton cropping in Xinjiang using high-throughput sequencing[J]. Archives of Microbiology, 2018, 200(4): 653-662. DOI:10.1007/s00203-018-1476-4 |
| [34] |
Mamtimin H, Yusuf K, Erkin R, et al. Temporal and spatial patterns of diversity and community structure of culturable endophytic bacteria isolated from Populus euphratica within the rivers of the Tarim basin[J]. Acta Ecologica Sinica, 2014, 34(22): 6622-6639. (in Chinese) 吾尔麦提汗·麦麦提明, 卡依尔·玉素甫, 热孜亚·艾肯, 等. 塔里木胡杨林可培养胡杨内生细菌多样性与群落结构的时空演变格局[J]. 生态学报, 2014, 34(22): 6622-6639. |
| [35] |
Mamtimin T, Kurban M, Hamdun M, et al. Cultivable bacterial diversity in natural Populus euphratica forest soils at the dried-up, ancient Kiyik River, Xinjiang, China[J]. Xinjiang Agricultural Sciences, 2016, 53(5): 921-934. (in Chinese) 吐尔逊阿依·麦麦提依明, 玛依古丽·库尔班, 马丽艳木·阿木东, 等. 新疆Kiyik废弃古河道天然胡杨林地可培养细菌的多样性研究[J]. 新疆农业科学, 2016, 53(5): 921-934. |
| [36] |
Rodrigues JLM, Pellizari VH, Mueller R, et al. Conversion of the Amazon rainforest to agriculture results in biotic homogenization of soil bacterial communities[J]. Proceedings of the National Academy of Sciences of the United States of America, 2013, 110(3): 988-993. DOI:10.1073/pnas.1220608110 |
| [37] |
Cheng ZB, Zhang FH, Gale WJ, et al. Effects of reclamation years on composition and diversity of soil bacterial communities in Northwest China[J]. Canadian Journal of Microbiology, 2018, 64(1): 28-40. DOI:10.1139/cjm-2017-0362 |
| [38] |
Li WJ, Tang SK, Wang D, et al. A primary study on biodiversity of moderately halophilic actinomycetes in Xinjiang and Qinghai Area[J]. Acta Microbiologica Sinica, 2004, 44(1): 1-7. (in Chinese) 李文均, 唐蜀昆, 王栋, 等. 新疆青海中度嗜盐放线菌生物多样性初步研究[J]. 微生物学报, 2004, 44(1): 1-7. DOI:10.3321/j.issn:0001-6209.2004.01.001 |
| [39] |
Xu LH, Li WJ, Cui XL, et al. Discovery of a vast amount of unknown actinomycetes from extreme environments in Xinjiang and Qinghai Province, China[J]. Journal of Yunnan University, 2003, 25(3): 283-292. |