 2021, Vol. 48
2021, Vol. 48扩展功能
文章信息
- 李强, 衣杨, 吴忠道, 丁涛
- LI Qiang, YI Yang, WU Zhongdao, DING Tao
- 基于机器学习的肠道菌群数据建模与分析研究综述
- Review of gut microbiome analysis prediction models and algorithms
- 微生物学通报, 2021, 48(1): 180-196
- Microbiology China, 2021, 48(1): 180-196
- DOI: 10.13344/j.microbiol.china.200346
-
文章历史
- 收稿日期: 2020-04-07
- 接受日期: 2020-06-12
- 网络首发日期: 2020-10-15





2. 中山大学新华学院信息科学学院 广东 广州 510520;
3. 中山大学中山医学院 广东 广州 510000
2. School of Information Science, Xinhua College of Sun Yat-Sen University, Guangzhou, Guangdong 510520, China;
3. Zhongshan School of Medicine, Sun Yat-Sen University, Guangzhou, Guangdong 510000, China
人体肠道内有1 000-1 150种细菌,数量达到约100万亿,是人体细胞数量的10倍,除此之外还含有少量的真菌、病毒和其他原生生物,这些微生物被统称为肠道菌群[1]。过去的研究已证实,肠道菌群在维持人体健康方面具有不可替代的作用,包括防御有害或致病菌的定殖[2],帮助消化食物并提供必要的维生素和营养素[3-6],以及维持一个健康的免疫系统[7]。与此同时,肠道菌群的扰动也会对人体的健康产生负面影响,这种影响称为肠道微生态失调[8],其可以导致多种疾病的发生,如肠易激综合征[9-10] (Irritable Bowel Syndrome,IBS)、自身免疫疾病[11]、2型糖尿病[12],以及疾病易感性增加,如癌症易感性[13]、肥胖易感性[14-15]等。目前普遍认为肠道菌群可作为反映人体健康状态以及疾病易感性的重要标志物,基于肠道菌群微生态系统建立疾病预测模型已成为目前一大新兴研究热点。
机器学习是一类能通过经验自动改进的计算机算法,其在解决聚类、分类、回归等问题中表现出独有的优势。随着近几年机器学习算法的不断优化,其在上述菌群数据分析与建模中发挥出独特优势,主要体现在菌群聚类分析[16-17]、菌群识别分类[18-21]和宿主表型预测[22-23]等方面。相较而言,利用机器学习构建疾病预测模型是一项更加复杂且更具挑战性的工作,经过众多科研人员的努力,近年来在该领域产生了许多令人欣喜的科研成果。
在构建疾病预测模型中,机器学习主要用作核心建模算法,其中包括基于支持向量机(Support Vector Machine,SVM)建模、基于随机森林(Random Forest,RF)建模、基于人工神经网络(Artificial Neural Network,ANN)建模等。如Ai等[24]利用RF构建了结直肠癌预测模型;Pasolli等[25]利用RF构建了2型糖尿病预测模型;Reiman等[20]使用ANN构建了肝硬化疾病预测模型;Wu等[26]使用KNN构建了2型糖尿病预测模型等。这些不同种类的疾病预测模型构建都是基于机器学习算法的自我学习能力与特定疾病数据集的结合,因此在特定数据集上具有很好的性能表现,但其普遍存在的问题是模型的泛化能力不足。
此外,部分科研人员也将机器学习作为数据处理模块用于疾病预测建模,主要包括数据降维(Data Dimension Reduction)、数据特征提取(Data Feature Detection)、主成分分析(Principal Component Analysis,PCA)等。如Montassier等[27]使用机器学习方法获取数据潜在特征,再结合统计学信息计算出患血液感染(Blood Stream Infection,BSI)疾病的风险指数,以及Oh等[28]通过深度学习处理宏基因组数据实现数据降维等。这些数据处理方法去除了冗余数据且能够在一定程度上提升算法的预测性能,但同时也增加了疾病预测模型的复杂程度,影响了整体的运行速率。
本文对机器学习算法在肠道菌群数据分析及疾病预测模型构建中的应用进行综述,梳理了肠道菌群数据分析手段的发展历程,重点归纳了应用机器学习方法构建疾病预测模型的具体步骤,进而阐述相关算法研究在疾病预测模型构建上的最新研究进展。本文以期为推动肠道菌群分析相关研究发展以及探索结合机器学习算法进行肠道菌群分析提供理论依据。同时,首次归纳整理出的疾病预测模型构建流程也可为开发基于肠道菌群数据的新型诊疗手段提供关键技术指导。
1 肠道菌群分析的历史进程及重要里程碑本文从肠道菌群数据分析的角度,对肠道菌群分析历史发展过程和其中的重要里程碑进行了相关梳理,将其分为3个阶段。
1.1 第一阶段(17世纪-20世纪末)在这一时期科研人员主要通过传统的生物学方法,在细胞层次对肠道菌群进行分析。人类肠道菌群研究始于17世纪,当时的列文虎克、莱迪、科赫等是研究宿主-微生物互作的先驱。1917年尼索首次分离出大肠杆菌益生菌菌株,明确了定殖抵抗的作用[29]。1944年Hungate发明了可培养厌氧菌的滚筒技术,成功分离培养了一种厌氧菌[30]。1958年Eiseman报道了将健康人类粪便以灌肠的方式进行粪菌移植(Fecal Microbiota Transplantation,FMT),成功医治了4例伪膜性肠炎病人[31]。1965年,Schaedler等首次将细菌移植给无菌小鼠,揭示了肠道菌群对宿主发育及生理功能的重要性,这一创举建立了利用无菌动物研究肠道菌群对宿主作用的新方法[32]。1989年,Janeway提出免疫细胞通过模式识别受体感知菌群信号以启动免疫应答,而在随后的研究中也进一步证实菌群对免疫系统具有调节作用[33]。
1.2 第二阶段(2000-2010年)随着测序技术的不断发展,利用测序信息对肠道菌群进行分析成为这一阶段的主要方式,肠道菌群分析进入分子主导阶段。2003年,Rowher首次利用宏基因组学方法在人类粪便中发现了此前未被培养的病毒组[34]。2005年,第二代测序技术的提出显著提升了16S rRNA基因扩增组的测序深度,可从分类层级上分析复杂菌群,同时也推动了利用宏基因组数据组装微生物基因组,帮助深入阐释人类微生态的功能与特征[35]。2007年,人类微生物组项目[36] (Human Microbiome Project,HMP)正式启动,标志着对于人类微生物菌群的研究从个体研究走向大规模人群的研究,自此大量微生物菌群数据开始产生。
1.3 第三阶段(2010年至今)由于肠道菌群研究中产生了大量测序数据,如何从海量数据中获取有用的知识成为研究人员的当务之急,此时机器学习的崛起为肠道菌群研究提供了新思路,肠道菌群分析也因此进入数据主导阶段。2010年,名为微生物生态学定量研究[37] (Quantitative Insights Into Microbial Ecology,QIIME)的工具开始使用,其能够对微生物组测序所产生的大量数据进行分析并解释。QIIME的出现,标志着机器学习方法开始正式用于进行微生物菌群分析。2016年,Pasolli等[25]提出基于肠道菌群数据使用机器学习构建疾病预测模型,这是肠道菌群机器分析发展过程中的一次重要尝试。事实证明,将机器学习用于预测模型的构建具有广阔前景。2018年,Asgari等[38]提出使用浅层k-mer代替OTU进行预测模型的构建,并首次提出使用深度学习来构建克罗恩病预测模型。2019年,Oh等[28]提出的DeepMicro方法,利用深度学习提取数据特征,再利用机器学习进行预测建模,实现了机器学习与深度学习结合运用于肠道菌群分析。Lo等[39]提出的MetaNN方法,基于肠道菌群数据,分别利用CNN和MLP构建预测模型,实现了对表型、身体部位以及疾病的准确预测。
2 人体肠道菌群分析常用的机器学习算法机器学习可以通过计算机在海量数据中学习数据的规律和模式,从中挖掘出潜在信息,广泛应用于解决分类、回归、聚类问题。根据训练数据是否预先分配标签可将其分为有监督学习和无监督学习,有标签则称为有监督学习,反之为无监督学习[40]。此外,当输出数据为离散值时称为分类问题,输出数据为连续值时则是回归问题[40]。
疾病预测建模是利用有标记的肠道菌群数据对机器学习模型进行训练,生成一个具备根据输入的肠道菌群数据判断宿主患病情况的预测模型,所以究其本质,疾病预测建模是一个有监督的分类问题。本文结合课题组在该领域的长期研究成果和近年来肠道菌群研究领域中疾病预测建模方面取得的进展,对常用的几种疾病预测建模算法进行分析和比较。
2.1 支持向量机(Support Vector Machine,SVM)SVM是一种有监督二分类器,当数据线性可分时,SVM通过在原始特征空间中构建一个最优分割超平面并将其作为决策面,最大化正负样本之间的边缘距离。当数据线性不可分时,SVM使用核函数将样本数据映射到一个高维空间,然后寻找一个最优分类超平面隔离不同类别样本数据,从而进行分类。
肠道菌群分析中,SVM因其良好的泛化能力以及在基于小样本构建而二分类模型上的独特优势,通常被作为一种基础算法被广泛应用,包括基于SVM构建菌群分类模型[41]、蛋白质预测模型[42]和疾病预测模型[43-44]等。然而,由于SVM对数据缺失这一问题极其敏感,因此在使用SVM进行肠道菌群数据建模时,通过进行数据预处理保证数据的完整性对于确保模型的性能至关重要。
2.2 K邻近(K Nearest Neighbors,KNN)算法KNN算法也是一种在肠道菌群分析中广泛应用的算法,该方法的基本原理是根据邻近样本来推断待测样本的类别。主要步骤包括:(1)计算每个测试样本和每个训练样本之间的距离。(2)找到距离最近的k个训练样本作为测试样本的最近邻居。(3)根据k个训练样本类别的众数作为测试样本进行分类。
相较于SVM,KNN算法的一大优势在于无需专门训练,而且更适合多分类问题。由于通常肠道菌群的数据样本量有限,无法进行大规模的模型训练,因此KNN算法非常适用于根据肠道菌群数据进行疾病预测的应用场景。如Wu等[26]使用了KNN构建2型糖尿病预测模型。然而正因为KNN算法没有模型训练过程,使其进行样本分类时的计算复杂度相对较高,而且容易受样本不均衡问题的影响。
2.3 随机森林(Random Forests,RF)算法RF算法是一种集成学习方法,通过自助法重采样技术,从原始训练样本集N中有放回地重复随机抽取n个样本生成新的训练样本集合训练决策树,然后按以上步骤生成m棵决策树组成随机森林,新数据的分类结果按分类树投票多少形成的分数而定。
RF算法具有训练速度快、适应不平衡数据集、适应多分类问题、泛化能力强等特性,适合进行肠道菌群数据分析。例如在处理16S rRNA基因等肠道菌群数据时,由于其天然可用来对分类问题中变量的重要性进行排序,使之能够在不做特征选择的情况下快速地进行模型训练,而且在处理肠道菌群数据不均衡的问题时可自动平衡样本误差。目前,RF算法已广泛应用于构建疾病预测模型,例如Ai等[24]利用RF构建结直肠癌预测模型;Pasolli等[25]利用RF构建2型糖尿病、肝硬化等疾病的预测模型。
2.4 人工神经网络(Annual Neural Network,ANN)算法ANN是一种模仿生物神经网络(动物的中枢神经系统,特别是大脑)结构和功能的计算模型,其具有自学习、自组织、自适应能力,主要包括多层感知机(Multiple Layer Perception,MLP)、卷积神经网络(Convolution Neural Network,CNN)、循环神经网络(Recursive Neural Network,RNN)、深度置信网络(Deep Belief Network,DBN)等。
ANN算法具备极强的从大量复杂数据中进行特征提取和特征表示的能力,通过构建人工神经网络对肠道菌群数据进行深度学习,能够挖掘出其中潜藏的深层次抽象性特征,从而构建分析性能更佳、泛化能力更强的数据模型。然而在肠道菌群数据分析中,使用ANN训练预测模型时,对数据量的要求将极大地提高,训练过程的控制(参数设置、迭代次数等)也将更加复杂,训练结果也会具有更大的未知性和不可解释性。因此,采用ANN方法进行建模,在具有明显优势的同时也带来诸多挑战。在以往使用ANN进行建模的研究中,Reiman等[20]尝试使用多层感知机、深度置信网络、卷积神经网络、递归神经网络分别构建了肝硬化疾病预测模型,并取得了较其他传统机器学习算法更好的多分类精度。
3 疾病预测模型构建流程基于肠道菌群数据构建的疾病预测模型中,模型构建过程是否合理规范直接决定了模型预测性能和预测结果的可靠性。本文通过分析近年来在肠道菌群数据分析领域的相关研究成果,结合本课题组在数据分析建模领域的长期积累,归纳总结了基于肠道菌群数据构建疾病预测模型的4个基本步骤(图 1):数据预处理、特征提取、算法选择、评估与验证。

|
| 图 1 疾病预测模型构建流程图 Figure 1 Flow chart of disease prediction model construction |
|
|
由于原始数据可能存在缺失值、样本不均衡、数据的形式不规范等问题,这会对模型的性能产生不利影响,严重的可能使得模型无法收敛,从而导致训练失败。在肠道菌群分析中,通过数据预处理过程对数据进行整理,从而使其满足模型训练要求,确保训练过程正常进行。常用的数据预处理操作包括了操作分类单元(Operate Taxonomy Unit,OTU)生成、样本均衡以及改变数据组织形式等。
3.1.1 OTU生成OTU生成是指对原始16S rRNA基因数据进行聚类操作,是菌群分析中最常见的预处理操作。聚类生成OTU主要有以下3种方法[45]:(1)无参OTU生成(de novo),是将所有序列直接按照两两之间的相似度进行聚类分析,划分成一个个OTU,选取该OTU中丰度最高的序列作为代表序列,然后用代表序列比对参考数据库,获得该OTU的物种注释。(2)有参OTU生成(Closed-Reference),这种方法是将序列与参考数据库直接比对,比对到同一参考序列的作为一个OTU,在OTU聚类的同时,也获得了该OTU的物种注释信息。(3)半有参OTU生成(Open-Reference),具有上述2种方法的特点,将序列与参考序列比对,未比对上的序列再进行无参OTU生成。
在完成OTU生成后,下一步需要将生成的OTU数据整合成OTU表,即经过OTU聚类和对OTU进行物种分类注释后生成的数据表格。在没有特殊分析要求时,整个OTU生成过程均可使用QIIME2[46]软件完成,而对于OTU表中可能存在的数据缺失问题,可采用均值插入、中位数插入等方法解决。
3.1.2 样本均衡现实场景中患病人群与健康人群在数量上存在很大差距,从而导致收集到的患病人员样例往往相对较少,因此,在根据肠道菌群数据构建模型的过程中容易出现样本不均衡问题。随机采样[47]是处理不均衡数据最基本的方法。该算法首先复制随机选择的少数类样本,并将生成的样本集合添加到少数类中,得到新的少数类集合。虽然其只是简单地将复制后的数据添加到原始数据集中,而且某些样本的多个实例都是“并列的”,但是也有可能使分类器学习出现过拟合现象[48]。为了有效解决随机采样算法的过拟合问题,科研人员尝试利用KNN方法来解决样本不均衡问题,如Batista等[49]提出一种SMOTE方法,这种方法首先寻找每一个少数类样本的k个同类最近邻样本,然后随机选择k个最近邻中的一个,并在这2个样本之间随机进行线性插值,构造出新的人工少数类样本。该方法可以有效地解决由于决策区间较小导致的分类过拟合问题,而且可使模型的学习能力得到显著提高。
3.1.3 数据组织形式改变肠道菌群分析中,改变数据组织形式是一种特殊的处理方式。通过这种方式能够实现将多种数据信息融合,从而为模型训练提供更加丰富的特征信息,以及使得组织后的数据形式能够适应某些有明确输入数据要求的训练模型,如卷积神经网络等。在Nguyen等[50]的研究中,将宏基因组数据投影到2D图像上,再将所生成的图像作为卷积神经网络的输入进行模型训练。此外,在Reiman等[20]的研究中,根据系统发育关系构建系统发育树,之后为树中的节点分配丰度,并在水平和垂直方向上使各节点保持相似距离。然后将系统发育树的节点关系映射到2D矩阵中,再用卷积神经网络进行模型训练。改变数据的组织形式,不仅提高了机器学习算法在进行肠道菌群数据分析时的适用性,也有助于发掘肠道菌群数据中的深层特征。
3.2 特征提取在基于肠道菌群数据构建预测模型时,利用特征提取手段获取丰富的数据特征是提升模型性能的重要手段。一般而言,特征提取的存在形式有2种:(1)特征提取作为单独一个步骤存在,按照研究人员的预先计划获取相应的特征信息,以这些信息作为模型训练数据进行训练。(2)将特征提取结合到模型训练阶段,通过这种方式获取的特征不会作为数据输出,而是直接由训练算法进行处理,无单独的特征提取阶段。本节主要讨论第一种特征提取形式。
根据特征提取的复杂程度可将特征提取的层级大致分为3个阶段:(1)对数据进行去噪和压缩处理,主要采用无监督学习的方法,如主成分分析、数据降维等。这一阶段获取的特征在基本保留原有数据形式的基础上,去除原有数据中的噪声和冗余信息,从而实现数据的简化和压缩。(2)收集数据的统计学特征,在这一阶段主要使用统计学方法,如统计OTU分类单元的相对丰度、α多样性、β多样性等。这一阶段获取的特征能反映数据的整体特点,因此往往与肠道微生态有一定联系。(3)提取数据的深层次抽象特征,其中主要包括两部分工作,一是通过机器学习的方法找到潜藏的特征信息标志量[26],如Montassier等[27]使用机器学习方法开发了一种BSI风险指数,该指数结合线性回归算法用于预测BSI发病率;二是采用深度学习方法将原始数据进行重组,例如Zhang等[17]通过浅层自编码器、深层自编码器、变分自编码器和卷积自编码器处理宏基因组数据,将高维数据转化为高鲁棒且富含特征信息的低维数据。
3.3 算法选择在构建疾病预测模型中,针对特定的问题场景(如数据特点等)、不同的性能需求(如准确率等),需要对比选取特定的机器学习算法。然而一直以来算法选取缺乏一般性准则,通常需要通过实验对比才能选出针对当前问题的最优算法。本文通过分析机器学习算法特点(图 2),结合这些算法在疾病预测建模中的性能表现,总结出以下关于疾病预测建模中选取机器学习算法的几点规律。

|
| 图 2 疾病预测建模中几种机器学习算法的性能对比 Figure 2 Performance comparison of several machine learning algorithms in disease prediction modeling |
|
|
(1) 根据数据特点选取建模算法。数据特点包括数据组织形式、数据完备性、数据冗余情况、数据均衡、数据量大小等。如数据存在较多缺失值时,则应优先选取对于数据缺失不敏感的算法,包括ANN、KNN等;如存在样本不均衡问题时可优先选取RF;如若数据量较少则应选择适合小样本训练的算法SVM。
(2) 根据模型性能需求选取建模算法。模型的性能需求包括模型处理的是二分类还是多分类问题,模型可解释性、模型处理速度、模型泛化能力等。如针对某一种疾病进行预测建模时,应选择适合处理二分类问题的算法包括SVM等;而在对多种疾病进行预测建模时,则应优先考虑ANN算法。
(3) 根据硬件配置选取建模算法。机器学习算法具有不同的时间复杂度和空间复杂度,对于空间复杂度要求高的算法如KNN、ANN,需要足够的计算机内存才能支撑模型的训练和运行;而对于时间复杂度高的算法如CNN、MLP,为了满足算法的性能要求则需要利用GPU来提升模型的运算速度。
此外,在长期的肠道菌群与疾病的研究中,科研人员陆续发现了多种与不同疾病相关联的标志菌。例如:在研究肠道菌群与肥胖的关联时,科研人员发现肥胖者体内富含大量产H2的普雷沃氏菌,采用实时定量PCR检测到利用H2的产甲烷古细菌在肥胖者体内明显多于正常体重的人[51];还有在研究肠道菌群与Ⅱ型糖尿病的关联时,科研人员发现Ⅱ型糖尿病以中度肠道微生物菌群失调为特征,一些常见的产丁酸盐细菌丰度下降,而各种致病菌增加,糖尿病人与非糖尿病人相比厚壁菌门相对丰度低,而拟杆菌门和变形菌门丰度高[52]。基于类似上述的研究成果,在利用机器学习构建疾病预测模型时,可优先选择易于添加先验条件的建模算法(如RF),将这些研究成果作为先验加入到疾病预测模型中,以此提升模型的可解释性和模型的预测性能。
总而言之,在选取建模算法时需要针对特定的疾病预测建模问题,综合考虑所选算法在多分类能力、可解释性、泛化能力、时空复杂度、数据容错性等方面的情况,并结合现有的研究成果,考虑算法在运用已有研究成果这一问题上的兼容性,选取合适的算法,使所选取的算法能够发挥其在构建特定疾病模型时的性能优势。
3.4 模型评估与验证模型评估与验证是整个模型构建中的最后一步,通过对模型的评估和验证能够得出所构建模型的相关性能指标,并判断模型是否具备实际运用的能力。
3.4.1 性能评估指标混淆矩阵(表 1)是衡量分类型模型准确度中最基本、最直观、计算最简单的方法,被广泛应用于基于肠道菌群的疾病预测模型的性能评估中,本文也以混淆矩阵为基础,介绍几种常用的性能评估指标。
| Predict | Actual (True) | Actual (False) |
| Predict (True) | TP (True positive) | FP (False positive) |
| Predict (False) | FN (False negative) | TN (True negative) |
(1) 正确率(Accuracy),表示所有预测样本中估计正确的样本个数。

|
(2) 错误率(Error Rate),表示所有预测样本中估计错误的样本个数。

|
(3) 准确率(Precision),表示所有判断为正的样本中实际样本为真的比例。
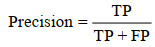
|
(4) 召回率(Recall),表示所有实际为真的样本中被估计为真的比例。
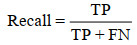
|
(5) F1分数(F1 Score),表示准确率和召回率的调和平均值。

|
(6) 操作者操作特征曲线(Receiver Operating Characteristic,ROC曲线)反映敏感性和特异性连续变量的综合指标,适用于二分类的情况,ROC曲线上的每个点反映着对同一信号刺激的感受性。横坐标是假正率(False Positive Rate),纵坐标是真正率(True Positive Rate),又称作召回率。
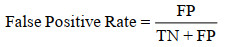
|
(7) AUC值(Area Under Curve),定义为ROC曲线下的面积,适用于二分类的情况,表示当随机挑选一个正样本以及一个负样本,当前的分类算法根据计算得到的Score值将这个正样本排在负样本前面的概率就是AUC值。当然,AUC值越大,当前的分类算法越有可能将正样本排在负样本前面,即能够更好地分类。
3.4.2 评估验证方法在运用机器学习进行疾病预测建模过程中,通常的做法是将数据分为训练集和测试集。测试集是与训练独立的数据,完全不参与训练,用于最终模型的评估。在训练过程中,经常会出现过拟合的问题。如果此时就使用测试数据来调整模型参数,就相当于在训练时已知部分测试数据的信息会影响最终评估结果的准确性。通常的做法是在训练数据中再分出一部分作为验证(Validation)数据,用来评估模型的训练效果。
验证数据取自训练数据,但不参与训练,这样可以相对客观地评估模型对于训练集之外数据的匹配程度。模型在验证数据中的评估常用的是K倍交叉验证,又称循环验证。其将原始数据分成K组(K-Fold),将每个子集数据分别做一次验证集,其余的K-1组子集数据作为训练集,这样会得到K个模型。这K个模型分别在验证集中评估结果,最后的误差MSE (Mean Squared Error)加和平均就得到交叉验证误差。交叉验证有效利用了有限的数据,并且评估结果能够尽可能地接近模型在测试集上的表现,是肠道菌群数据建模中的主要验证手段。
4 疾病预测建模典型实例过去的研究已证实,机器学习在解决肠道菌群分类识别的问题上表现出了很好的性能,随着研究的深入,许多科研团队开始探索将机器学习用于构建基于肠道菌群数据的疾病预测模型,根据所采用算法的特点可将其大致分为两类:(1)基于传统机器学习算法的预测模型;(2)基于深度学习算法的预测模型。
4.1 基于传统机器学习的预测模型在基于机器学习构建的预测模型的发展历程中,最早采用传统的机器学习算法进行模型构建,这些算法包括SVM、RF、KNN等。这类算法普遍的特点是简便易操作、处理速度快。
在2016年,Pasolli等[25]提出了MetAML (Metagenomic Rediction Analysis Based on Machine Learning)预测工具,利用MetAML构建单一疾病预测模型的基本步骤包括(图 3A):(1)对宏基因组数据按照人类微生物组项目的标准操作规程进行预处理,并删除某些仅含少量核苷酸的Reads (如:对于IBD数据集最小核苷酸个数为70)。(2)采用默认参数的MetaPhlAn2[53]方法进行特征提取,获得物种水平的相对丰度和特异菌株标记两种特征。(3)分别基于SVM、RF等算法建立预测模型。(4)利用10倍交叉验证对所构建模型的性能进行评估测试,结果见表 2。该模型只能预测一种疾病发病情况(由训练数据集决定),本质上是一个二分类模型,因此模型的适用性较差,而其优势在于预测精度高、模型构建过程简单。

|
| 图 3 典型实例建模流程 Figure 3 Typical example modeling process |
|
|
| Algorithm | RF | RF | SVM | SVM |
| Performance index | F1 score | AUC | F1 score | AUC |
| Cirrhosis disease | 0.88 | 0.945 | 0.83 | 0.922 |
| Colorectal cancer | 0.79 | 0.873 | 0.73 | 0.809 |
| IBD | 0.75 | 0.89 | 0.78 | 0.862 |
| T2D | 0.66 | 0.744 | 0.61 | 0.663 |
| WT2D | 0.69 | 0.762 | 0.57 | 0.664 |
此外,在研究[25]中还发现,模型预测能力通常是通过针对性的特征选择和使用特定于菌株的标记来提高的,而不是通过物种级别的分类丰度来提高;而且通过在某一疾病训练样本中添加其他疾病训练集中健康样本,能够提高针对该疾病预测模型的预测能力。
2018年,Wu等[26]开发出一种可以表征多标签样本的方法,并运用该方法构建了一个可以同时预测多种疾病的模型(包括肝硬化、2型糖尿病、风湿性关节炎);构建该模型的基本步骤包括(图 3B):(1)利用BWA (Burrows Wheeler Aligner) “MEM”模块进行数据预处理,与此同时,采用Near Miss和SMOTEEN方法进行上采样以克服样本不均衡问题。(2)构建丰度矩阵,计算关联丰度并采用mRMR方法根据最大相关最小冗余原则选取前500个特征。(3)将这500个特征的数据利用KNN、LR、RF、SVM、GBDT等算法分别进行建模。(4)采用10倍交叉验证对所构建模型性能进行评估测试,结果见表 3。该模型最大的优势是可以同时预测多种疾病,但其局限性则是预测的准确率会因此下降。
| Performance index | SVM | RF | KNN |
| F1 score (Based Near Miss) | 0.718 4 | 0.788 8 | 0.662 8 |
| F1 score (Based SMOTEENN) | 0.913 8 | 0.834 1 | 0.860 2 |
同时,Wu等[26]还在构建基于肠道菌群数据的疾病预测模型的过程中发现了多种疾病的微生物标记信息,例如:T2D患者的拟杆菌门/厚壁菌门比值发生明显改变;在肝硬化患者中发现了丰富的链球菌和细孔菌,特别是另一种名为粪杆菌的细菌。以上发现不仅再次验证了人体菌群的复杂性和对于不同疾病的特异性,也进一步表明将机器学习用于肠道菌群数据分析是具有重要意义的研究课题。
在构建疾病预测模型时,除了将传统机器学习算法作为预测模型的主体外,有的科研团队也采取将机器学习作为辅助手段再结合其他模型的方式对疾病进行预测。如Montassier等[27]使用机器学习方法对16S rRNA基因序列进行处理,再结合多个统计学数据计算出BSI风险指数,该指数仅基于预处理粪便微生物组用以预测BSI发病率。此外,还有针对疾病治疗的预后反应建立预测模型,如Ananthakrishnan等[54]利用人工神经网络结合临床和微生物相关数据开发了一个综合预测模型,用以预测疾病治疗的反应。
4.2 基于深度学习的算法的预测模型在大数据时代,将肠道菌群大数据转化为有价值的知识是肠道菌群研究中面临的重要挑战之一。深度学习是机器学习的分支,其特点是具有强大的学习能力和灵活性。近年来,深度学习得到了迅速发展,在各个领域都展现出了巨大的性能优势。
2018年,Asgari等[38]使用浅层k-mer代替OTU进行预测模型的构建,并首次提出利用深度学习构建克罗恩病预测模型(图 3C),其基本原理是:(1)首先将16S rRNA基因序列经过自举阶段处理,得到一个合适的样本大小N,使其可以代表整个数据并产生稳定的k-mer谱。(2)利用亚采样序列寻找k的最佳值进行分类,生成样本的k-mer表示。(3)将样本的k-mer表示作为训练数据,并采用多层感知机(Mutiple Layer Perception,MLP)进行预测模型构建。(4)使用10倍交叉验证评估算法性能,结果见表 4。在该研究中,Asgari等[38]还将使用深度学习构建的模型与传统机器学习模型进行了比较,根据比较结果得知,在面对简单的二分类问题时,传统机器学习算法的性能与深度学习的性能相当,但在处理多分类时,深度学习所构建模型的性能明显优于传统机器学习所构建的模型。
| Feature | Algorithm | Precision | Recall | F1 score |
| 6-Mer feature | RF | 0.76±0.04 | 0.76±0.04 | 0.76±0.04 |
| 6-Mer feature | SVM | 0.68±0.04 | 0.68±0.04 | 0.68±0.04 |
| OTU feature | RF | 0.74±0.04 | 0.74±0.04 | 0.74±0.04 |
| OTU feature | SVM | 0.68±0.04 | 0.68±0.04 | 0.68±0.04 |
其次,在文献[38]中,作者选择了浅层k-mer而非OTU数据进行深度学习,并对使用k-mer进行数据建模的优势进行总结,包括:(1)利用k-mer表示肠道菌群数据易于计算机处理,并能有效节约计算开销。(2)当样本包含未知分类时,与分类无关的分析通常是扩增子测序的首选方法,而k-mer特征正好满足这一点,因为其不需要对分类做任何假设。(3) k-mer分布是一种定义良好的肠道菌群数据特征表示方法,当采用OTU表示肠道菌群数据时则存在对通道和参数敏感等问题。(4)测序序列的相似性通过k-mer表示被自然地引入到后续的深度学习算法,而当将序列分组到某些类别时,OTU之间的序列相似性被忽略。在后续的对比实验中,也证实基于k-mer表示肠道菌群数据特征的方法的确在训练速度和预测精度上相比OTU表示有一定提升。
除了上述的单独采用深度学习进行疾病预测建模之外,还有研究人员提出将传统机器学习与深度学习相结合共同构建疾病预测模型。如Oh等[28]提出的DeepMicro方法(图 3D),该方法通过浅层自编码器、深层自编码器、变分自编码器和卷积自编码器来处理微生物菌群的宏基因组数据,将高维数据转化为高鲁棒的低维数据,再将处理好的数据交由其他机器学习的算法(如SVM、RF、MLP)进行疾病预测建模。该方法将深度学习算法在发掘深层特征上的优势和传统机器学习算法在解决二分类问题上的优势相结合,增加了数据的特征,从而使得模型泛化能力提高;该模型在炎症性肠病、欧洲和中国人群的2型糖尿病、肝硬化和肥胖症这5个数据集上进行实验,结果(表 5)表明以这种结合方式建模在预测性能上均优于单独的算法建模;此外,作者还对DeepMicro构建的模型与未使用DeepMicro构建的模型在运行效率上进行了比较,根据实验结果(表 6),基于DeepMicro构建的模型在运算速率上快5-30倍[28]。
| Prediction model | Based DeepMicro | Best of now |
| Performance index | AUC | AUC |
| Cirrhosis disease | 0.940 | 0.945 |
| Colorectal cancer | 0.803 | 0.811 |
| IBD | 0.955 | 0.921 |
| T2D | 0.763 | 0.750 |
| WT2D | 0.899 | 0.832 |
| Prediction model | Normal | Based DeepMicro | |||||
| SVM | RF | MLP | SVM | RF | MLP | ||
| Cirrhosis disease | 777 | 72 | 4 508 | 17 | 40 | 287 | |
| Colorectal cancer | 187 | 50 | 4 508 | 2 | 30 | 105 | |
| IBD | 126 | 42 | 3 776 | 2 | 28 | 103 | |
| T2D | 1 705 | 99 | 12 057 | 8 | 47 | 188 | |
| WT2D | 85 | 41 | 2 449 | 2 | 28 | 93 | |
本文选取Cirrhosis Disease、Colorectal Cancer、IBD、T2D和WT2D这5种疾病的数据集分别在DeepMicro预测模型和MetAML预测模型上的预测性能进行横向比较,结果见图 4。

|
| 图 4 DeepMicro与MetAML性能(AUC值)对比结果 Figure 4 Performance comparison between DeepMicro and MetAML |
|
|
通过横向比较可得出以下分析结果:(1)基于DeepMicro构建的模型在性能上优于基于MetAML构建的模型。(2)基于DeepMicro构建的模型性能稳定性更高,预测性能基本维持在较高水平,而基于MetAML构建的模型性能稳定性较低,会因为数据集的不同而产生较大的变化。(3)存在个别数据集上(如Colorectal Cancer) MetAML构建的模型性能优于DeepMicro构建的模型。
上述分析中DeepMicro是基于深度学习的方法,MetAML是基于传统机器学习的方法,所以该比较结果一定程度上体现了深度学习在肠道菌群数据分析领域的优势。由于在传统机器学习算法中,特征工程主要是基于相关理论研究人为设置,这种特征工程方法的优势在于具有较强的理论基础和可解释性,但也因此限制了数据的表现能力,很多潜在特征无法被利用,因而造成了当某一特征适合于当前疾病预测时,基于该特征构建的模型对该疾病会有较好的预测性能,反之则可能造成模型预测性能不佳的情况。然而,通过采用深度学习的方法,一方面可以扩增特征的维度,提升其表现能力,另一方面也发掘出了其中的深层次特征,能够找到对多种疾病均适用的普适性特征,从而使其在整体预测性能和泛化能力上均优于传统机器学习方法。
5 人体肠道菌群基准测试数据集 5.1 16S rRNA基因测序数据16S rRNA基因是细菌染色体上编码rRNA相对应的DNA序列,存在于所有细菌的染色体基因组中,由于其高信息量、高保守型及与大多数生理、遗传标记一致,已经广泛应用于微生物物种间进化和微生物多样性研究[55]。此外,随着第二代测序技术的发展,16S rRNA基因数据的获取成本已明显降低,对菌群的16S rRNA基因序列进行数据分析已成为肠道菌群研究中的主要手段(表 7)。
| Dataset | Region | Normal case | Disease case | Samples | Data type | References |
| Cirrhosis disease dataset | Intestinal | 83 | 98 | 181 | 16S rRNA gene | [56] |
| Colorectal cancer dataset | Intestinal | 61 | 95 | 156 | 16S rRNA gene | [57] |
| IBD dataset | Intestinal | 124 | 16S rRNA gene | [58] | ||
| T2D dataset | Intestinal | 74 | 71 | 145 | 16S rRNA gene | [59] |
| WT2d Dataset | Intestinal | 43 | 102 | 145 | 16S rRNA gene | [60] |
| Human microbiome project (first stage) | Intestinal, Skin, Oral, etc | 300 | 16S rRNA gene and metagenome | [36] | ||
| Human microbiome project (second stage) | Intestinal, Skin, Oral, etc | 16S rRNA gene and metagenome | [62] |
(1) 肝硬化数据集[56]。该数据集是由Qin等[56]针对肝硬化疾病收集整理而成。作者从98名肝硬化患者和83名健康对照者的粪便样本中提取总体DNA库,利用Illumina HiSeq 2000进行测序。每个样品平均产生4.74 Gb的高质量序列,总共得到860 Gb的16S rRNA基因序列数据。
(2) 结直肠癌数据集[57]。2014年Zeller等[57]采集156名参与者(包含了53名结直肠癌患者、42名肿瘤患者和61名随机健康对照组)的粪便样本,之后在Illumina HiSeq 2000/2500平台上对收集的粪便样本进行全基因组鸟枪法测序,并将所有测序样本以100 bp的读取长度进行配对末端测序,从而得到结直肠癌数据集。
(3) 炎症性肠病数据集[58]。Qin等[58]采集124名志愿者的粪便样本,对所有样本用不同的克隆插入尺寸构建配对端文库,并进行Illumina GA测序。所有Reads均使用Soapnovo19进行组装。通过对所有基因的两两比较,在一致性为95%、重叠率为90%的条件下,利用BLAT36构建炎症性肠病非冗余基因集。
(4) 2型糖尿病数据集[59]。Qin等[59]采集145名参与者(包含71例2型糖尿病患者和74例健康对照者)的粪便样本,并利用全基因组测序方法获取样本中的菌群DNA数据,之后对所有DNA样本进行测序,平均每个样本获得2.61 Gb配对端Reads,获得总计378.4 Gb的高质量DNA数据。
(5) 女性2型糖尿病数据集[60]。Karlsson等[60]使用鸟枪法测序分析145名欧洲妇女(包括53名T2D患者、49名糖耐量受损和43名健康个体)的粪便样本全基因组序列,用标准程序从粪便样本中提取基因组DNA,并在Illumina HiSeq 2000上进行测序,每个样本平均获得3.1±1.8 Gb的测序数据,共计约449 Gb数据。
5.2 宏基因组数据相较于单一的16S rRNA基因序列数据,宏基因组数据包括了群落中菌群的全部遗传信息,无疑能更好地表征环境微生物样本。分析宏基因组数据既可以获得微生物群落的组成信息,包括物种的组成和丰度等,又能获得较全面的功能信息。例如,微生物蛋白质编码基因、生物代谢反应中关键酶的表达,甚至更详尽的代谢反应网络。较高质量的宏基因组数据还可以从中提取出特定的核酸序列,如有算法提出可以从宏基因组数据中提取出16S rRNA基因序列数据,并能较好地避免扩增偏差[61] (表 7)。
(1)人类微生物组计划(第1阶段)数据集[36]:人类微生物组计划由美国国立卫生局提出并于2008年正式启动,该项目的第一阶段描述了来自300个健康个体的微生物群落,它们分布在人体的几个不同部位:鼻腔、口腔、皮肤、胃肠道和泌尿生殖道。通过16S rRNA基因测序来描述每个身体部位微生物群落的复杂性,采用宏基因组全基因组鸟枪测序为了解人类微生物群落的功能和途径提供了思路。总共产生了超过14.23 Tb的数据,主要提供3种类型的数据:参考微生物基因组,宏基因组鸟枪测序序列,16S rRNA基因序列。
(2) 人类微生物组计划(第2阶段)数据集[62]:2014年成立的综合人体微生物组项目是美国国家卫生研究院共同基金人类微生物组项目的第2阶段,这一阶段使用多种“组学”技术从微生物组和宿主的微生物学相关条件的3个不同队列研究(怀孕和早产,炎症性肠病的发病,2型糖尿病的发病)中创建生物学特性的整合纵向数据集,该数据集包括了16S rRNA基因序列数据和宏基因组鸟枪测序数据。
6 总结与展望近年来,宏基因组研究的迅速兴起,现有的方法正变得越来越复杂,测序技术正取得指数级的进步。如Illumina平台引入了更适合小基因组测序的仪器,具有更快的运行时间和更长的读取长度,为宏基因组应用提供了更大的灵活性。牛津纳米孔公司(Oxford Nanopore)开发的技术可以在可扩展的系统中实现长时间读取和短时间运行,因此非常适合微生物应用。以上技术的发展,极大地简便了获取肠道菌群测序数据的流程,推动了肠道菌群分析的发展。此外,大量的微生物组研究项目如人类微生物组、人肠道宏基因组学(Metagenomics of the Human Intestinal Tract,MetaHIT)相继启动,产生了大量的数据。这些数据为理解微生物菌群与人体之间的复杂联系创造了巨大的机会,同时也带来了巨大的计算和理论挑战,大致集中于:(1)提取微生物数据集并进行识别分类。(2)微生物组数据降维和可视化。(3)利用机器学习方法推断微生物和疾病之间的相关性。(4)模拟微生物群落在人体内的动态发展过程。
本文从肠道菌群数据分析的角度入手,主要针对基于肠道菌群数据构建疾病预测模型这一问题,梳理了肠道菌群研究的发展历程和重要里程碑;根据过往的研究工作,整理出肠道菌群数据分析中常用的7个基准数据集;总结出基于肠道菌群数据构建疾病预测模型的基本流程;重点阐述了近年来传统机器学习和深度学习在构建疾病预测模型中的典型应用。
目前机器学习方法在肠道菌群分析领域已广泛应用,奠定了利用肠道菌群预测疾病发生发展进程中的技术基础,有力推动了基于肠道菌群的新型诊疗手段的发展。然而,基于本课题组在采用机器学习算法进行数据建模方面的长期研究和实践发现[63-64],相较于机器学习算法在模式分类和其他发展相对成熟的领域,目前采用机器学习算法进行肠道菌群数据分析的研究还存在以下不足:(1)在将机器学习算法运用于肠道菌分析时仅停留在应用层面,而缺少对算法的特异性优化和对机器学习算法本身的研究。(2)肠道菌群的数据表示形式单一且存在大量的冗余信息,无法为机器学习提供足够的数据信息。(3)大量研究中采用的数据集规模较小,基本集中在200-300人,而肠道菌群具有丰富的多样性,因而所构建模型的泛化能力不足。
此外,本课题组还长期致力于深度学习领域的相关研究,通过大量数据进行深度学习,挖掘出其中潜藏的深层特征,并以此构建具备解决复杂分析问题能力的数据分析模型,而这对肠道菌群数据分析具有重要借鉴意义。例如,在本课题组以往的实践[65]中,采用深度学习方法构建了基于大量教育统计数据的线上教育管理系统,通过对大数据的深层次分析,解决了线上教育管理困难的问题。同样,在面对大量的肠道菌群原始数据时,由于无法准确地针对特定问题选取相应的数据特征,因而可以考虑对肠道菌群数据进行深度学习,挖掘其中与问题相关的潜在特征,再基于此构建数据模型解决目标问题。同时,在课题组以往的科研过程中利用卷积神经网络在图像和视频领域进行过大量的研究工作,其关键优势在于能够发掘二维数据中的结构化特征和潜在共性,从而进行分类和识别,例如利用卷积神经网络有效地发掘出图像信息中隐字[66],以及利用卷积神经网络实现对图像特定目标的识别、跟踪[67-69]以及对视频中人体行为进行识别[70-71]。在对肠道菌群数据进行分析建模时,也可以通过对肠道菌群数据的组织形式进行适当调整,如将系统进化树组织成二维数据的形式,将各种丰度信息用图像形式表示等,探索将卷积神经网络用于肠道菌群数据分析的方式。
近年来,图卷积神经网络取得了突破性的进展,其在处理图数据的分类和预测时展现出了强大的性能优势,目前在交通预测、人体行为识别、生物分类等领域均取得了诸多成果。在肠道菌群研究中,系统进化生成树的结构以及菌群之间的关联均可通过图结构进行表示,那么是否可以考虑将肠道菌群分析与图神经网络进行结合,利用图卷积特性分析肠道菌群数据,以此推动肠道菌群分析领域的研究和发展。
综上所述,在未来,机器学习和深度学习方法应该被视为分析肠道菌群的一个重要工具,我们应该积极探索这二者间恰当的结合点,借助计算机科学上的研究成果推动肠道菌群分析的发展,实现将肠道菌群的分析作为临床上诊断、治疗和预防疾病的有力工具。
| [1] |
Guo HL, Shao YY, Menghe BLG, Zhang HP. Research on the relation between gastrointestinal microbiota and disease[J]. Microbiology China, 2015, 42(2): 400-410. (in Chinese) 郭慧玲, 邵玉宇, 孟和毕力格, 张和平. 肠道菌群与疾病关系的研究进展[J]. 微生物学通报, 2015, 42(2): 400-410. |
| [2] |
Walsh CJ, Guinane CM, O'Toole PW, Cotter PD. Beneficial modulation of the gut microbiota[J]. FEBS Letters, 2014, 588(22): 4120-4130. DOI:10.1016/j.febslet.2014.03.035 |
| [3] |
Ramakrishna BS. Role of the gut microbiota in human nutrition and metabolism[J]. Journal of Gastroenterology and Hepatology, 2013, 28(S4): 9-17. |
| [4] |
Hennessy AA, Ross RP, Fitzgerald GF, Caplice N, Stanton C. Role of the gut in modulating lipoprotein metabolism[J]. Current Cardiology Reports, 2014, 16(8): 515. DOI:10.1007/s11886-014-0515-2 |
| [5] |
Fuller M. Determination of protein and amino acid digestibility in foods including implications of gut microbial amino acid synthesis[J]. British Journal of Nutrition, 2012, 108(S2): S238-S246. DOI:10.1017/S0007114512002279 |
| [6] |
Dutton RJ, Turnbaugh PJ. Taking a metagenomic view of human nutrition[J]. Current Opinion in Clinical Nutrition & Metabolic Care, 2012, 15(5): 448-454. |
| [7] |
Cantorna MT, McDaniel K, Bora S, Chen J, James J. Vitamin D, immune regulation, the microbiota, and inflammatory bowel disease[J]. Experimental Biology and Medicine, 2014, 239(11): 1524-1530. DOI:10.1177/1535370214523890 |
| [8] |
Tamboli CP, Neut C, Desreumaux P, Colombel JF. Dysbiosis in inflammatory bowel disease[J]. Gut, 2004, 53(1): 1-4. DOI:10.1136/gut.53.1.1 |
| [9] |
Kostic AD, Xavier RJ, Gevers D. The microbiome in inflammatory bowel disease: current status and the future ahead[J]. Gastroenterology, 2014, 146(6): 1489-1499. DOI:10.1053/j.gastro.2014.02.009 |
| [10] |
Cammarota G, Ianiro G, Cianci R, Bibbò S, Gasbarrini A, Currò D. The involvement of gut microbiota in inflammatory bowel disease pathogenesis: potential for therapy[J]. Pharmacology & Therapeutics, 2015, 149: 191-212. |
| [11] |
Collado MC, Rautava S, Isolauri E, Salminen S. Gut microbiota: a source of novel tools to reduce the risk of human disease?[J]. Pediatric Research, 2015, 77(1/2): 182-188. |
| [12] |
Larsen N, Vogensen FK, van den Berg FWJ, Nielsen DS, Andreasen AS, Pedersen BK, Al-Soud WA, Sørensen SJ, Hansen LH, Jakobsen M. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults[J]. PLoS One, 2010, 5(2): e9085. DOI:10.1371/journal.pone.0009085 |
| [13] |
Viaud S, Daillère R, Boneca IG, Lepage P, Pittet MJ, Ghiringhelli F, Trinchieri G, Goldszmid R, Zitvogel L. Harnessing the intestinal microbiome for optimal therapeutic immunomodulation[J]. Cancer Research, 2014, 74(16): 4217-4221. DOI:10.1158/0008-5472.CAN-14-0987 |
| [14] |
Cox LM, Blaser MJ. Antibiotics in early life and obesity[J]. Nature Reviews Endocrinology, 2015, 11(3): 182-190. DOI:10.1038/nrendo.2014.210 |
| [15] |
Gohir W, Ratcliffe EM, Sloboda DM. Of the bugs that shape us: maternal obesity, the gut microbiome, and long-term disease risk[J]. Pediatric Research, 2015, 77(1/2): 196-204. |
| [16] |
Soueidan H, Nikolski M. Machine learning for metagenomics: methods and tools[J]. arXiv preprint arXiv: 1510.06621, 2015
|
| [17] |
Zhang Y, Hu XH, Jiang XP. Multi-view clustering of microbiome samples by robust similarity network fusion and spectral clustering[J]. IEEE/ACM Transactions on Computational Biology and Bioinformatics, 2017, 14(2): 264-271. DOI:10.1109/TCBB.2015.2474387 |
| [18] |
Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy[J]. Applied and Environmental Microbiology, 2007, 73(16): 5261-5267. DOI:10.1128/AEM.00062-07 |
| [19] |
Oudah M, Henschel A. Taxonomy-aware feature engineering for microbiome classification[J]. BMC bioinformatics, 2018, 19(1): 1-13. DOI:10.1186/s12859-017-2006-0 |
| [20] |
Reiman D, Metwally A, Dai Y. Using convolutional neural networks to explore the microbiome[A]//2017 39th Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC)[C]. Seogwipo, South Korea: IEEE, 2017: 4269-4272
|
| [21] |
Zieliński B, Plichta A, Misztal K, Spurek P, Brzychczy-Włoch M, Ochońska D. Deep learning approach to bacterial colony classification[J]. PLoS One, 2017, 12(9): e0184554. DOI:10.1371/journal.pone.0184554 |
| [22] |
Johnson HR, Trinidad DD, Guzman S, Khan Z, Parziale JV, DeBruyn JM, Lents NH. A machine learning approach for using the postmortem skin microbiome to estimate the postmortem interval[J]. PLoS One, 2016, 11(12): e0167370. DOI:10.1371/journal.pone.0167370 |
| [23] |
Thompson J, Johansen R, Dunbar J, Munsky B. Machine learning to predict microbial community functions: an analysis of dissolved organic carbon from litter decomposition[J]. PLoS One, 2019, 14(7): e0215502. DOI:10.1371/journal.pone.0215502 |
| [24] |
Ai LY, Tian HY, Chen ZF, Chen HM, Xu J, Fang JY. Systematic evaluation of supervised classifiers for fecal microbiota-based prediction of colorectal cancer[J]. Oncotarget, 2017, 8(6): 9546-9556. DOI:10.18632/oncotarget.14488 |
| [25] |
Pasolli E, Truong DT, Malik F, Waldron L, Segata N. Machine learning meta-analysis of large metagenomic datasets: tools and biological insights[J]. PLoS Computational Biology, 2016, 12(7): e1004977. DOI:10.1371/journal.pcbi.1004977 |
| [26] |
Wu HL, Cai LH, Li DF, Wang XY, Zhao SC, Zou FH, Zhou K. Metagenomics biomarkers selected for prediction of three different diseases in Chinese population[J]. BioMed Research International, 2018, 2018: 2936257. |
| [27] |
Montassier E, Al-Ghalith GA, Ward T, Corvec S, Gastinne T, Potel G, Moreau P, de la Cochetiere MF, Batard E, Knights D. Pretreatment gut microbiome predicts chemotherapy-related bloodstream infection[J]. Genome Medicine, 2016, 8(1): 49. DOI:10.1186/s13073-016-0301-4 |
| [28] |
Oh M, Zhang LQ. DeepMicro: deep representation learning for disease prediction based on microbiome data[J]. Scientific Reports, 2020, 10(1): 6026. DOI:10.1038/s41598-020-63159-5 |
| [29] |
Wehkamp J, Harder J, Wehkamp K, Wehkamp-von Meissner B, Schlee M, Enders C, Sonnenborn U, Nuding S, Bengmark S, Fellermann K, et al. NF-κB and AP-1-mediated induction of human beta defensin-2 in intestinal epithelial cells by Escherichia coli Nissle 1917: a novel effect of a probiotic bacterium[J]. Infection and Immunity, 2004, 72(10): 5750-5758. DOI:10.1128/IAI.72.10.5750-5758.2004 |
| [30] |
Hungate RE. Studies on cellulose fermentation: I. The culture and physiology of an anaerobic cellulose-digesting bacterium[J]. Journal of Bacteriology, 1944, 48(5): 499-513. DOI:10.1128/JB.48.5.499-513.1944 |
| [31] |
Kassam Z, Lee CH, Yuan Y, Hunt RH. Fecal microbiota transplantation for Clostridium difficile infection: Systematic review and meta-analysis[J]. The American Journal of Gastroenterology, 2013, 108(4): 500-508. DOI:10.1038/ajg.2013.59 |
| [32] |
Schaedler RW, Dubos R, Costello R. The development of the bacterial flora in the gastrointestinal tract of mice[J]. The Journal of Experimental Medicine, 1965, 122(1): 59-66. DOI:10.1084/jem.122.1.59 |
| [33] |
Janeway CA Jr. Approaching the asymptote? Evolution and revolution in immunology[A]//Cold Spring Harbor Symposia on Quantitative Biology[C]. New York: Cold Spring Harbor Laboratory Press, 1989, 54: 1-13
|
| [34] |
Rohwer F. Global phage diversity[J]. Cell, 2003, 113(2): 141. DOI:10.1016/S0092-8674(03)00276-9 |
| [35] |
Liu C, Li JB, Rui JP, An JX, Li XZ. The applications of the 16S rRNA gene in microbial ecology: current situation and problems[J]. Acta Ecologica Sinica, 2015, 35(9): 2769-2788. (in Chinese) 刘驰, 李家宝, 芮俊鹏, 安家兴, 李香真. 16S rRNA基因在微生物生态学中的应用[J]. 生态学报, 2015, 35(9): 2769-2788. |
| [36] |
The Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome[J]. Nature, 2012, 486(7402): 207-214. DOI:10.1038/nature11234 |
| [37] |
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, et al. QⅡME allows analysis of high-throughput community sequencing data[J]. Nature Methods, 2010, 7(5): 335-336. DOI:10.1038/nmeth.f.303 |
| [38] |
Asgari E, Garakani K, McHardy AC, Mofrad MRK. MicroPheno: predicting environments and host phenotypes from 16S rRNA gene sequencing using a k-mer based representation of shallow sub-samples[J]. Bioinformatics, 2018, 34(13): i32-i42. DOI:10.1093/bioinformatics/bty296 |
| [39] |
Lo C, Marculescu R. MetaNN: accurate classification of host phenotypes from metagenomic data using neural networks[J]. BMC Bioinformatics, 2019, 20(S12): 314. DOI:10.1186/s12859-019-2833-2 |
| [40] |
Zhou ZH. Machine Learning[M]. Beijing: Tsinghua University Press, 2016. (in Chinese) 周志华. 机器学习[M]. 北京: 清华大学出版社, 2016. |
| [41] |
Kung HC, Chen RM, Tsai JJP, Hu RM. Stratification of human gut microiome and building a SVM-based classifier[A]//2018 IEEE 18th International Conference on Bioinformatics and Bioengineering (BIBE)[C]. Taichung, Taiwan, China: IEEE, 2018: 14-17
|
| [42] |
Xu L, Liang GM, Liao CR, Chen GD, Chang CC. An efficient classifier for Alzheimer's disease genes identification[J]. Molecules, 2018, 23(12): 3140. DOI:10.3390/molecules23123140 |
| [43] |
Chen HM, Yu YN, Wang JL, Kong X, Yang CQ, Yang L, Liu ZJ, Yuan YZ, Fei L, et al. Decreased dietary fiber intake and structural alteration of gut microbiota in patients with advanced colorectal adenoma[J]. The American of Clinical Nutrition, 2013, 97(5): 1044-1052. DOI:10.3945/ajcn.112.046607 |
| [44] |
Larsen PE, Dai Y. Metabolome of human gut microbiome is predictive of host dysbiosis[J]. Gigascience, 2015, 4(1): s13742-015-0084-3. DOI:10.1186/s13742-015-0084-3 |
| [45] |
Navas-Molina JA, Peralta-Sánchez JM, González A, McMurdie PJ, Vázquez-Baeza Y, Xu ZJ, Ursell LK, Lauber C, Zhou HW, Song SJ, et al. Advancing our understanding of the human microbiome using QⅡME[A]//Methods in Enzymology[M]. San Diego, CA: Academic Press, 2013, 531: 371-444
|
| [46] |
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Alghalith GA, Alexander H, Arumugam M, Bai Y, et al. Reproducible, interactive, scalable and extensible microbiome data science using QⅡME 2[J]. Nature biotechnology, 2019, 37(8): 852-857. DOI:10.1038/s41587-019-0209-9 |
| [47] |
Libbrecht MW, Noble WS. Machine learning applications in genetics and genomics[J]. Nature Reviews Genetics, 2015, 16(6): 321-332. DOI:10.1038/nrg3920 |
| [48] |
Tao XM, Hao SY, Zhang DX, Xu P. Overview of classification algorithms for unbalanced data[J]. Journal of Chongqing University of Posts and Telecommunications (Natural Science Edition), 2013, 25(1): 101-110. (in Chinese) 陶新民, 郝思媛, 张冬雪, 徐鹏. 不均衡数据分类算法的综述[J]. 重庆邮电大学学报:自然科学版, 2013, 25(1): 101-110. |
| [49] |
Batista GE, Prati RC, Monard MC. A study of the behavior of several methods for balancing machine learning training data[J]. ACM SIGKDD Explorations Newsletter, 2004, 6(1): 20-29. DOI:10.1145/1007730.1007735 |
| [50] |
Nguyen TH, Chevaleyre Y, Prifti E, Sokolovska N, Zucker JD. Deep learning for metagenomic data: using 2D embeddings and convolutional neural networks[Z]. arXiv preprint arXiv: 1712.00244, 2017
|
| [51] |
Zackular JP, Baxter NT, Iverson KD, Sadler WD, Petrosino JF, Chen GY, Schloss PD. The gut microbiome modulates colon tumorigenesis[J]. mBio, 2013, 4(6): e00692-13. |
| [52] |
Zhao LP, Zhang F, Ding XY, Wu GJ, Lam YY, Wang XJ, Fu HQ, Xue XH, Lu CH, Ma JL, et al. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes[J]. Science, 2018, 359(6380): 1151-1156. DOI:10.1126/science.aao5774 |
| [53] |
Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, Tett A, Huttenhower C, Segata N. MetaPhlAn2 for enhanced metagenomic taxonomic profiling[J]. Nature Methods, 2015, 12(10): 902-903. DOI:10.1038/nmeth.3589 |
| [54] |
Ananthakrishnan AN, Luo CW, Yajnik V, Khalili H, Garber JJ, Stevens BW, Cleland T, Xavier RJ. Gut microbiome function predicts response to anti-integrin biologic therapy in inflammatory bowel diseases[J]. Cell Host & Microbe, 2017, 21(5): 603-610.e3. |
| [55] |
Zhou C, Zhang SW, Chen W. Comparison of the clustering algorithms based on operational taxonomic units[J]. Beijing Biomedical Engineering, 2014, 33(6): 591-597. (in Chinese) 周晨, 张绍武, 陈伟. 微生物分类单元聚类算法比较研究[J]. 北京生物医学工程, 2014, 33(6): 591-597. DOI:10.3969/j.issn.1002-3208.2014.06.07 |
| [56] |
Qin N, Yang FL, Li A, Prifti E, Chen JF, Shao L, Guo J, Le Chatelier E, Yao J, Wu LJ, et al. Alterations of the human gut microbiome in liver cirrhosis[J]. Nature, 2014, 513(7516): 59-64. DOI:10.1038/nature13568 |
| [57] |
Zeller G, Tap J, Voigt AY, Sunagawa S, Kultima JR, Costea PI, Amiot A, Böhm J, Brunetti F, Habermann N, et al. Potential of fecal microbiota for early-stage detection of colorectal cancer[J]. Molecular Systems Biology, 2014, 10(11): 766. DOI:10.15252/msb.20145645 |
| [58] |
Qin JJ, Li RQ, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. A human gut microbial gene catalogue established by metagenomic sequencing[J]. Nature, 2010, 464(7285): 59-65. DOI:10.1038/nature08821 |
| [59] |
Qin JJ, Li YR, Cai ZM, Li SH, Zhu JF, Zhang F, Liang SS, Zhang WW, Guan YL, Shen DQ, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes[J]. Nature, 2012, 490(7418): 55-60. DOI:10.1038/nature11450 |
| [60] |
Karlsson FH, Tremaroli V, Nookaew I, Bergström G, Behre CJ, Fagerberg B, Nielsen J, Bäckhed F. Gut metagenome in European women with normal, impaired and diabetic glucose control[J]. Nature, 2013, 498(7452): 99-103. DOI:10.1038/nature12198 |
| [61] |
Moitinho-Silva L, Steinert G, Nielsen S, Hardoim CCP, Wu YC, McCormack GP, López-Legenti S, Marchant R, Webster N, Thomas T, et al. Predicting the HMA-LMA status in marine sponges by machine learning[J]. Frontiers in Microbiology, 2017, 8: 752. DOI:10.3389/fmicb.2017.00752 |
| [62] |
Integrative HMP (iHMP) Research Network Consortium. The integrative human microbiome project[J]. Nature, 2019, 569(7758): 641-648. DOI:10.1038/s41586-019-1238-8 |
| [63] |
Xu J, Chang HY, Xu C, Yi Y. Novel shared-path protection algorithm and reliability analysis model based on Bayesian network for multilink failures[J]. Optical Engineering, 2009, 48(12): 125002. DOI:10.1117/1.3275451 |
| [64] |
Yi Y, Wu JS, Xu W. Incremental SVM based on reserved set for network intrusion detection[J]. Expert Systems with Applications, 2011, 38(6): 7698-7707. DOI:10.1016/j.eswa.2010.12.141 |
| [65] |
Yi Y, Zhao ZH, Zhang J, Chen YH, Pan ZH, Hou JX, Su Y. 39: CN 108615423A[P]. 2018-10-02 (in Chinese) 衣杨, 赵泽慧, 张俊, 陈怡华, 潘志宏, 候景贤, 苏堉.一种基于深度学习的线上教育管理系统: 中国, 108615423A[P]. 2018-10-02 |
| [66] |
Ye J, Ni JQ, Yi Y. Deep learning hierarchical representations for image steganalysis[J]. IEEE Transactions on Information Forensics and Security, 2017, 12(11): 2545-2557. DOI:10.1109/TIFS.2017.2710946 |
| [67] |
Yi Y, Mo ZW, Tan JW. A novel hierarchical data association with dynamic viewpoint model for multiple targets tracking[J]. Journal of Visual Communication and Image Representation, 2016, 34: 37-49. DOI:10.1016/j.jvcir.2015.10.010 |
| [68] |
Yi Y, Cheng Y, Xu CP. Visual tracking based on hierarchical framework and sparse representation[J]. Multimedia Tools and Applications, 2018, 77(13): 16267-16289. DOI:10.1007/s11042-017-5198-4 |
| [69] |
Zheng ZX, Yi Y, Shen JL, Zhang JH. Adaptive updating siamese network with like-hood estimation for surveillance video object tracking[A]//2019 IEEE International Conference on Multimedia & Expo Workshops (ICMEW)[C]. Shanghai, China: IEEE, 2019: 126-131
|
| [70] |
Yi Y, Hu P, Deng XK. Human action recognition with salient trajectories and multiple kernel learning[J]. Multimedia Tools and Applications, 2018, 77(14): 17709-17730. DOI:10.1007/s11042-017-5209-5 |
| [71] |
Yi Y, Zheng ZX, Lin MQ. Realistic action recognition with salient foreground trajectories[J]. Expert Systems with Applications, 2017, 75: 44-55. DOI:10.1016/j.eswa.2017.01.008 |