 2020, Vol. 47
2020, Vol. 47扩展功能
文章信息
- 吴悦妮, 冯凯, 厉舒祯, 王朱珺, 张照婧, 邓晔
- WU Yue-Ni, FENG Kai, LI Shu-Zhen, WANG Zhu-Jun, ZHANG Zhao-Jing, DENG Ye
- 16S/18S/ITS扩增子高通量测序引物的生物信息学评估和改进
- In-silico evaluation and improvement on 16S/18S/ITS primers for amplicon high-throughput sequencing
- 微生物学通报, 2020, 47(9): 2897-2912
- Microbiology China, 2020, 47(9): 2897-2912
- DOI: 10.13344/j.microbiol.china.200054
-
文章历史
- 收稿日期: 2020-01-17
- 接受日期: 2020-04-12
- 网络首发日期: 2020-05-09





2. 中国科学院大学资源与环境学院 北京 100049;
3. 大连理工大学环境学院 工业生态与环境工程教育部重点实验室 辽宁 大连 116024;
4. 山东大学海洋研究院 山东 青岛 266237
2. College of Resources and Environment, University of Chinese Academy of Sciences, Beijing 100049, China;
3. Key Laboratory of Industrial Ecology and Environmental Engineering, Ministry of Education; School of Environmental Science and Technology, Dalian University of Technology, Dalian, Liaoning 116024, China;
4. Institute of Marine Science and Technology, Shandong University, Qingdao, Shandong 266237, China
扩增子测序是一种高靶向性地分析特定基因组区域中基因变异的方法,是发现单核苷酸多态性(single nucleotide polymorphisms,SNPs)的理想方法。其利用PCR的引物来扩增基因组的特定区域,靶向地捕获目标区域的DNA,达到目的DNA片段的富集目标;最后针对扩增产物(也被称为扩增子)进行高通量测序,分析序列中的遗传变异等信息。一般认为,核糖体RNA (rRNA)基因广泛存在于所有细胞生物体中,由于很少发生大规模的横向基因迁移,因此具有一系列由非常保守到高变的区域,适合于微生物分类信息的确定。由于核糖体操纵子具有足够的保守性,因此常被用作多样性调查的标记基因[1],主要包括16S rRNA、18S rRNA和转录间隔区(internal transcribed spacer,ITS)等。用下一代测序技术(next generation sequencing,NGS)测定16S/18S/ITS某个可变区的序列,可以反映环境样品在细菌、真菌、古菌等组成的微生物群落之间的差异,对研究海洋、土壤、宿主等不同环境中的微生物群落组成及动态变化有重要的指导作用,同时也是系统发育和分类学研究中一种广泛使用的方法[2]。
原核生物(细菌、古菌) rRNA按核酸的沉降系数分为3种,分别为5S、16S和23S。16S rRNA是核糖体RNA的一个亚基,16S rRNA基因就是编码该亚基的基因。其存在于所有细菌染色体基因组中,参与生物蛋白质的合成过程,并在生物进化的漫长历程中保持一定的遗传保守性,是细菌系统分类研究中最有用和最常用的分子标记[3]。16S rRNA基因分子大小适中(长度约为1 540 bp),在结构与功能上又具有高度的保守性,检测方便并且与原核生物的系统发育关联密切,因此逐渐成为最常用的原核生物标记基因。16S rRNA编码基因序列共有9个保守区和9个高可变区(图 1),其中V4-V6区数据库信息全、特异性好,是细菌多样性分析的常用目标区域[4]。

|
| 图 1 8对原核生物16S rRNA基因扩增引物及其相对位置 Figure 1 Eight primers and their positions for 16S rRNA gene amplification in prokaryotes |
|
|
在真核微生物中,核糖体DNA是由核糖体基因及与之相邻的间隔区组成,其基因组序列从5′到3′依次为:外部转录间隔区(external transcribed spacer,ETS)、18S rRNA基因、内部转录间隔区1 (ITS1)、5.8S rRNA基因、内部转录间隔区2 (ITS2)、28S rRNA基因和基因间隔序列(intergenic spacer,IGS) (图 2)。其中,18S rRNA基因是编码真核生物核糖体小亚基的DNA序列,其中既有保守区,也有可变区(V1-V9)。与16S rRNA基因相似,保守区反映了生物物种间的亲缘关系,而可变区则能体现物种间的差异,适用于作为物种分类单元及系统发育的分子标记。在真菌中,相比于rRNA中的18S、5.8S和28S的基因组序列,内转录间隔区ITS1和ITS2作为非编码区具有更高的丰富度和分辨率,承受的选择压力较小,相对变化较大,并且能够提供详尽的系统学分析所需要的可遗传性状[5]。同时,ITS的保守型表现为种内相对一致,种间差异较明显,能够反映出种属间甚至菌株间的差异。此外,ITS序列片段较小(ITS1和ITS2长度分别为350 bp和400 bp左右,但不同物种之间存在一定差异),易于分析。也就是说,ITS具有更高的扩增和测序成功率以及分类学分辨率,目前已被广泛应用于真菌不同种属的系统发育分析。

|
| 图 2 真核微生物核糖体DNA结构示意图 Figure 2 Structural diagram of eukaryotic microbial ribosomal DNA |
|
|
在过去的10年中,NGS技术被广泛用于探索各种生态系统中微生物群落的多样性和组成结构[6],诸如全球气候变化及微生物响应[7-9]、生物地理分布[10-11]、海洋生态系统[12]、农业生态系统[13]、生物降解及修复[14]、野生动物[15]及人体共生微生物[16]等各个领域。随着高通量测序技术的不断发展和参考数据库的不断更新,利用核糖体操纵子作为DNA标记可以揭示不同生态系统中的微生物多样性和组成[17]。然而,针对不同环境样本的引物选择和实验过程仍然需要更细致的验证[18-20]。本文收集了微生物群落中针对细菌、古菌、真菌所常用的标记基因扩增引物,包括16S rRNA基因(细菌和古菌)、ITS基因(真菌)和18S rRNA基因(真核微生物),并用不同数据库对其进行了生物信息学的评价,以期对不同环境样本和研究对象的扩增引物选择能起到一定的指导意义。
1 材料与方法 1.1 16S rRNA基因扩增引物与数据库我们收集了16S rRNA基因扩增常用的8对通用引物(图 1),其中包括4条正向引物:27F[21]、355F[22]、515F[23]、1114F[24];以及7条反向引物:342R[25]、519R[26]、806R[27]、926R[28]、1064R[22]、1392R[29]、1492R[30]。针对不同的扩增区域,关注了扩增V1-V2区的27F/342R引物对,扩增V3区的成对引物355F/519R引物对,扩增V3-V5区的成对引物355F/806R引物对,扩增V4-V5区的成对引物515F/806R引物对,扩增V4-V6区的成对引物515F/926R引物对,扩增V4-V7区的成对引物515F/1064R引物对,扩增V9区的成对引物1114F/1392R引物对,以及扩增16S rRNA基因全长(V1-V10区)的成对引物27F/1492R引物对(表 1)。
| No. | Primers name | Sequences (5'→3') | Amplified region | Amplified fragment length (bp) |
| 1 | 27F | AGRGTTYGATYMTGGCTCAG | V1-V2 | 315 |
| 342R | CTGCTGCSYCCCGTAG | |||
| 2 | 355F | ACWCCTACGGGWGGCWGC | V3 | 164 |
| 519R | GWATTACCGCGGCKGCTG | |||
| 3 | 355F | ACWCCTACGGGWGGCWGC | V3-V5 | 451 |
| 806R | GGACTACNVGGGTWTCTAAT | |||
| 4 | 515F | GTGYCAGCMGCCGCGGTAA | V4-V5 | 291 |
| 806R | GGACTACNVGGGTWTCTAAT | |||
| 5 | 515F | GTGYCAGCMGCCGCGGTAA | V4-V6 | 411 |
| 926R | CCGYCAATTYMTTTRAGTTT | |||
| 6 | 515F | GTGYCAGCMGCCGCGGTAA | V4-V7 | 550 |
| 1064R | AYCTCACGRCACGAGCTGAC | |||
| 7 | 1114F | GCAACGAGCGCAACCC | V9 | 278 |
| 1392R | ACGGGCGGTGTGTRC | |||
| 8 | 27F | AGRGTTYGATYMTGGCTCAG | V1-V10 | 1 465 |
| 1492R | TACGGYTACCTTGTTACGACTT |
选用SILVA数据库(SILVA123_QIIME_release,2015)对以上16S rRNA基因通用引物进行覆盖度的评价。SILVA是一个rRNA基因序列的综合数据库,收录了原核和真核微生物的小亚基rRNA基因序列(简称SSU,即16S和18S rRNA)和大亚基rRNA基因序列(简称LSU,即23S和28S rRNA)。细菌和真菌的都有,能够提供定期的更新和全面优质的检查。选用SILVA的16S rRNA基因数据库来评价16S通用引物,数据库包含原核生物16S rRNA基因序列226 267条。为了只保留数据库中16S全长序列,以确保每个可变区扩增引物的覆盖度评价结果更具有可比性,删除了数据库中序列长度小于1 500 bp的序列,保留了19 084条长度在1 500 bp以上的序列,构建为“SILVA 16S rRNA基因全长序列数据库”(表 2)。
| Gene name | Database | Number of sequences |
| 16S rRNA gene | SILVA 16S rRNA基因全长序列数据库 SILVA 16S rRNA gene full-length sequence database |
19 084 |
| SILVA 16S rRNA基因古菌序列数据库 SILVA 16S rRNA gene archaeal sequence database |
9 825 | |
| SILVA 16S rRNA基因非古菌序列数据库 SILVA 16S rRNA gene non-archaeal sequence database |
216 442 | |
| ITS gene | Unite ITS1基因序列数据库 Unite ITS1 gene sequence database |
3 970 |
| Unite ITS2基因序列数据库 Unite ITS2 gene sequence database |
11 367 | |
| Unite ITS基因全长序列数据库 Unite ITS gene full-length sequence database |
1 871 | |
| 海洋真菌ITS1基因序列数据库 Marine ITS1 gene sequence database |
683 | |
| 海洋真菌ITS2基因序列数据库 Marine ITS2 gene sequence database |
1 281 | |
| 海洋真菌ITS基因全长序列数据库 Marine ITS gene full-length sequence database |
113 | |
| 18S rRNA gene | SILVA 18S rRNA基因真核微生物数据库 SILVA 18S rRNA gene database for eukaryotic microbes |
25 497 |
| SILVA 18S rRNA基因真菌序列数据库 SILVA 18S rRNA gene fungal sequence database |
3 321 | |
| SILVA 18S rRNA基因非真菌序列数据库 SILVA 18S rRNA gene non-fungal sequence database |
22 176 | |
| 海洋真菌18S rRNA基因序列数据库 Marine 18S rRNA gene sequence database |
1 116 |
此外还收集了用来扩增古菌16S rRNA基因的正向引物A519F[31]及其改进版A519F*[32],反向引物A806R[33]和A908R[34]。其中,A519F和A806R的扩增区域为V4-V5区,A519F*和A908R的扩增区域为V4-V6区(表 3)。为了评价古菌16S rRNA基因特异引物,从SILVA的16S rRNA基因数据库中分离出古菌序列共9 825条,构建了“SILVA 16S rRNA基因古菌序列数据库”用来评价古菌16S rRNA基因扩增引物的覆盖度。同时,用去除古菌序列之后SILVA 16S rRNA数据库中剩余的216 442条序列构成了“SILVA 16S rRNA基因非古菌序列数据库”,用该数据库的引物覆盖度来评价古菌引物对于古菌序列的特异性。由于这两对引物的扩增区域均位于16S rRNA基因中部,因此未对数据库序列长度进行筛选。扩增16S rRNA基因V4-V5区的通用引物对515F/806R和扩增V4-V6区的通用引物对515F/926R也一起用来评价古菌引物的相对覆盖度与特异性。
| Primers name | Sequences (5′→3′) | Amplified region | Amplified fragment length (bp) |
| A519F | CAGCCGCCGCGGTAA | V4–V5 | 287 |
| A806R | GGACTACNSGGGTMTCTAAT | ||
| A519F* | CAGCMGCCGCGGTAA | V4–V6 | 389 |
| A908R | CCCGCCAATTCCTTTAAGTT |
收集了9对用于扩增ITS基因的通用引物(表 4),包括7条正向引物:ITS1[35]、ITS1F[36]、gITS7[37]、gITS7ngs[38]、5.8S-Fun[39]、ITS3_KYO2[40]、ITS9MUNng[41];以及4条反向引物:ITS2[35]、ITS4R[35]、ITS4ngs[42]、ITS4-Fun[39]。针对不同的扩增区域关注了9对引物对,其中包括扩增ITS1区的2对引物对ITS1/ITS2和ITS1F/ITS2,扩增ITS2区的4对引物对gITS7/ITS4R、gITS7ngs/ITS4ngs、5.8S-Fun/ITS4-Fun和ITS3_KYO2/ITS4R,扩增ITS全长序列的3对引物对ITS1/ITS4R、ITS1F/ITS4R和ITS9MUNngs/ITS4ngs。
| No. | Primers name | Sequence (5′→3′) | Position | Amplified region | Amplified fragment length (bp) |
| 1 | ITS1 | TCCGTAGGTGAACCTGCGG | 18S | ITS1 | 220 |
| ITS2 | GCTGCGTTCTTCATCGATGC | 5.8S | |||
| 2 | ITS1F | CTTGGTCATTTAGAGGAAGTAA | 18S | 190 | |
| ITS2 | GCTGCGTTCTTCATCGATGC | 5.8S | |||
| 3 | gITS7 | GTGARTCATCGARTCTTTG | 5.8S | ITS2 | 315 |
| ITS4R | TCCTCCGCTTATTGATATGC | 28S | |||
| 4 | gITS7ngs | GTGARTCATCRARTYTTTG | 5.8S | 315 | |
| ITS4ngs | TCCTSCGCTTATTGATATGC | 28S | |||
| 5 | 5.8S-Fun | AACTTTYRRCAAYGGATCWCT | 5.8S | 325 | |
| ITS4-Fun | (AG)CCTCCGCTTATTGATATGCTTAART | 28S | |||
| 6 | ITS3_KYO2 | GATGAAGAACGYAGYRAA | 5.8S | 360 | |
| ITS4R | TCCTCCGCTTATTGATATGC | 28S | |||
| 7 | ITS1 | TCCGTAGGTGAACCTGCGG | 18S | ITS1+5.8S+ITS2 | 510 |
| ITS4R | TCCTCCGCTTATTGATATGC | 28S | |||
| 8 | ITS1F | CTTGGTCATTTAGAGGAAGTAA | 18S | 540 | |
| ITS4R | TCCTCCGCTTATTGATATGC | 28S | |||
| 9 | ITS9MUNngs | TTGTACACACCGCCCGTCG | 18S | 650 | |
| ITS4ngs | TCCTSCGCTTATTGATATGC | 28S |
由于真核微生物的核糖体DNA是由核糖体基因及与之相邻的间隔区(ITS)组成(图 2),使扩增ITS1的正向引物落在18S亚基上,扩增ITS1的反向引物和扩增ITS2的正向引物落在5.8S,而扩增ITS2的反向引物则是落在28S亚基上。然而目前并没有一个较为准确完整的数据库能够提供从18S至28S全长序列。因此,我们用ITSx Extractor从专门针对真菌ITS序列的数据库Unite中分别筛选出的ITS1、ITS2和ITS全长序列[43],并构建了3个新的数据库。其中,ITS1数据库的筛选条件为:包含ITS1基因,且ITS1基因前、IT1基因后序列长度 > 100 bp;ITS2数据库的筛选条件为:包含ITS2基因,且ITS2基因前、ITS2基因后序列长度 > 100 bp;ITS基因全长序列数据库的筛选条件为:序列包含ITS1基因,ITS2基因、且ITS1基因前、ITS1基因与ITS2基因间隔、ITS2基因后序列长度 > 100 bp。基于此,筛选出ITS1序列3 970条、ITS2序列11 367条、ITS全长序列1 871条,并以此分别构建了“Unite ITS1基因序列数据库” “Unite ITS2基因序列数据库”和“Unite ITS基因全长序列数据库”,用于评价ITS通用引物的覆盖度(表 2)。此外,还从GenBank中收集了海洋真菌的ITS序列15 468条(关键词:Marine、Ocean、Costal、Estuary),并用上述方法筛选出ITS1序列683条、ITS2序列1 281条以及ITS全长序列113条,基于此构建了“海洋真菌ITS1基因序列数据库” “海洋真菌ITS2基因序列数据库”和“海洋真菌ITS基因全长序列数据库”,用于判断通用引物对海洋环境的适用性(表 2)。
1.3 18S rRNA基因扩增引物与数据库对于18S rRNA基因,收集了5对引物(表 5),包括4条正向引物:SSU_F_22[44]、TAReuk454FWD1[45]、nu-SSU-0817[46]、Euk1391f[47];以及5条反向引物:SSU_R_09[44]、SSU_R_13[44]、R1200[48]、nu-SSU- 1196[46]、EukBr[47]。针对不同的扩增区域关注了扩增V3区的引物对SSU_F_22/SSU_R_09、扩增V3-V7区的引物对SSU_F_22/SSU_R_13、扩增V4-V5区的引物对TAReuk454FWD1/R1200、扩增V5区的引物对nu-SSU-0817/nu-SSU-1196以及扩增V9区的引物对Euk1391f/EukBr。其中,nu-SSU-0817/nu-SSU-1196为真菌特异性引物对,其他4对引物为真核微生物通用引物对。由于Euk1391f/EukBr的扩增片段是18S rRNA基因最末端的一个可变区V9区,反向引物EukBr落在了ITS1基因上,其他引物则均位于18S rRNA基因的保守区(表 5)。
| No. | Primers name | Sequences (5'→3') | Amplified fragment length (bp) | Amplified region |
| 1 | SSU_F_22 | TCCAAGGAAGGCAGCAGGC | 137 | V3 |
| SSU_R_09 | AGCTGGAATTACCGCGGCTG | |||
| 2 | SSU_F_22 | TCCAAGGAAGGCAGCAGGC | 990 | V3–V7 |
| SSU_R_13 | GGGCATCACAGACCTGTTA | |||
| 3 | TAReuk454FWD1 | CCAGCASCYGCGGTAATTCC | 600 | V4–V5 |
| R1200 | CCCGTGTTGAGTCAAATTAAGC | |||
| 4 | nu-SSU-0817 | TTAGCATGGAATAATRRAATAGGA | 385 | V5 |
| nu-SSU-1196 | TCTGGACCTGGTGAGTTTCC | |||
| 5 | Euk1391f | GTACACACCGCCCGTC | 130 | V9 |
| EukBr* | TGATCCTTCYGCAGGTTCACCTAC |
我们选择SILVA 18S rRNA基因真核微生物数据库来评价以上通用引物,数据库共包含25 497条真核微生物18S rRNA基因序列,且序列长度均 > 900 bp。其中包括真菌序列3 321条,基于此我们构建了“SILVA 18S rRNA基因真菌序列数据库”,剩余序列则构建为“SILVA 18S rRNA基因非真菌序列数据库”,包括非真菌序列22 176条(表 2)。由于扩增V9区的反向引物EukBr落在ITS1基因上,而并没有一个较为完整的数据库可以包含18S+ITS1全长序列,因此我们用Unite ITS1基因序列数据库来另外评价反向引物EukBr的覆盖度(表 2)。跟ITS基因一样,我们从GenBank收集了海洋真菌的18S rRNA基因序列13 459条(关键词:Marine、Ocean、Costal、Estuary)。由于本研究关注的引物中最长的扩增长度为1 200 bp,因此从该数据库中筛选出长度 > 1 300 bp的序列1 116条,基于此构建了“海洋真菌18S rRNA基因序列数据库”,用于判断通用引物对海洋环境的适用性。海洋真菌ITS1基因序列数据库也用来另外评价反向引物EukBr对于海洋样本的覆盖度(表 2)。
1.4 覆盖度与特异性计算引物覆盖度的评价方法选用了BLASTn序列比对。引物与数据库序列进行比对,并通过比对结果来计算每条引物和成对引物在数据库序列的覆盖度。计算方法如下:
用集合S表示一个有N条序列的数据库,那么S={S1, S2, S3, ……, S4},N = |S|,其中|S|是集合S的模,表示集合中序列的个数;假定简并引物P有m种可能性,分别记为Pi(i 1, 2, ……, m),Pi与数据库中的序列进行比对后,覆盖到的序列可以构成一个集合,记为S(Pi);
那么,上述S(Pi)的并集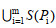
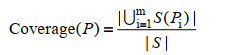
|
记P的反向引物为P',相似的,P'的覆盖度为

|
记P-P'为P和P'这一对引物,那么,P-P'在数据库S中覆盖序列的集合,就是正向引物P和反向引物P'在数据库S中覆盖序列的交集,即:
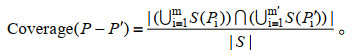
|
本研究收集和整理了SILVA数据库中19 084条长度在1 500 bp以上的序列,构建为16S全长序列数据库。该数据库共包含69个细菌和古菌门(phylum),2 202个可鉴定的属(genus),5 105个可鉴定的种(species) (图 3)。基于此,我们系统地评价了在16S rRNA基因的高通量测序中最常使用的8对通用引物,其中包括4条正向引物和7条反向引物(表 1)。在不允许碱基错配和仅允许一个碱基错配的情况下,分别计算了这些引物对于SILVA 16S rRNA全长序列数据库的覆盖度。

|
| 图 3 SILVA 16S rRNA基因全长序列数据库物种组成(门分类单元) Figure 3 The composition of SILVA database for 16S rRNA gene full-length sequences in phylum level |
|
|
在不允许错配的情况下,这11条引物中除了27F、1114F和1492R三条引物之外,其他引物的覆盖度均在80%以上。其中355F、515F、926R和1392R四条引物的覆盖度达到了90%以上,806R和1064R的覆盖度也达到了85%以上。从引物对来看,V4-V6区515F/926R引物对的评价结果最好,覆盖度能达到87.05%,V3-V5区355F/806R引物对、V4-V5区515F/806R引物对、V4-V7区515F/1064R引物对的覆盖度也均达到80%以上(图 4)。在允许一个碱基错配的情况下,引物的覆盖度几乎都得到了较大的提升。除了27F、342R、1114F和1492R四条引物之外,其他引物的覆盖度均在90%以上,515F、519R和1392R三条引物的覆盖度达到了95%以上。从引物对来看,V3区引物对355F/519R和V4-V6区引物对515F/926R的评价结果最好,覆盖度能达到91%以上。V3-V5区引物对355F/806R、V4-V5区引物对515F/806R、V4-V7区引物对515F/1064R的覆盖度也均达到87%以上(图 4)。

|
| 图 4 16S rRNA基因扩增引物在SILVA 16S rRNA基因全长序列数据库中的覆盖度 Figure 4 Coverage of 16S rRNA gene amplification primers in SILVA 16S rRNA gene full-length sequence database |
|
|
采用构建的SILVA 16S rRNA基因古菌序列数据库和SILVA 16S rRNA基因非古菌序列数据库(表 2),分别评价了古菌16S rRNA基因扩增引物对于古菌序列的覆盖度和特异性。图 5结果表明,A519F作为扩增古菌16S rRNA基因的最常用正向引物,有着较好的覆盖度和特异性,该引物可以覆盖到SILVA 16S rRNA基因数据库中高达90.46%的古菌序列和仅仅匹配0.03%的其他细菌序列。之后的研究中,研究者对A519F引物进行了改进,即A519F*引物,原本的第5个碱基C改为简并碱基M,加入了A的可能性[32]。然而从A519F和A519F*引物的对比可以看出,简并碱基的加入虽然略微提高了覆盖度(92.52%),但是却大大降低了针对古菌的特异性,而对细菌的覆盖度也大大提高了,对细菌序列的覆盖度高达93.32%。另外,反向引物A806R和A908R均表现出较高的覆盖度和较好的特异性。在不允许碱基错配和允许1个碱基错配的情况下,A806R对古菌序列的覆盖度分别可以达到87.60%和94.63%,特异性为11.57%。A908R的覆盖度相对低一点,在不允许碱基错配和允许1个碱基错配的情况下,分别可以达到82.78%和90.44%,但特异性评价结果更好,仅能覆盖数据库中不到0.01%的非古菌序列。

|
| 图 5 16S rRNA基因古菌扩增引物在SILVA 16S rRNA基因古菌序列数据库中的覆盖度及其对古菌的特异性 Figure 5 Coverage of 16S rRNA gene archaea amplification primers in SILVA 16S rRNA gene archaea database and their specificity to archaea |
|
|
为了评价古菌引物的相对覆盖度与特异性,扩增16S rRNA基因V4-V5区的通用引物对515F/806R和扩增V4-V6区的通用引物对515F/926R也分别用SILVA 16S rRNA基因古菌序列数据库和SILVA 16S rRNA基因非古菌序列数据库进行了覆盖度以及对古菌序列特异性的评价。结果显示,通用引物对于古菌序列的覆盖度也都很高(图 5)。在不允许碱基错配的情况下,古菌引物对于古菌序列的覆盖度均略高于通用引物;但在允许1个碱基错配的情况下,通用引物的覆盖度反而更高。也就是说,这两对通用引物都能较好地反映群落细菌与古菌组成。
2.2 ITS基因通用引物评价结果通过收集和整理Unite数据库中的真菌ITS基因序列,重新构建了Unite ITS1基因序列数据库、Unite ITS2基因序列数据库和Unite ITS基因全长序列数据库(表 2),并以此为基础系统地评价了在ITS基因的高通量测序中常使用的9对通用引物,包括扩增ITS1区的2对引物、扩增ITS2区的4对引物和扩增ITS全长序列的3对引物(表 4)。其中,3条正向引物ITS1、ITS1F和ITS9MUNngs位于18S亚基上;1条反向引物ITS2以及4条正向引物gITS7、gITS7ngs、5.8S-Fun和ITS3_KYO2位于5.8S亚基上;3条反向引物ITS4R、ITS4ngs和ITS4-Fun位于28S亚基上。在不允许碱基错配和仅允许1个碱基错配的情况下,分别计算了这些引物对于Unite 3个ITS序列数据库的覆盖度。此外,分别基于海洋真菌ITS1基因序列数据库、海洋真菌ITS2基因序列数据库和海洋真菌ITS基因全长序列数据库,系统评价了ITS通用引物对海洋真菌序列的普适性。
对于ITS1的扩增主要关注了2条正向引物ITS1和ITS1F以及1条反向引物ITS2 (图 6A)。正向引物ITS1对于Unite数据库和海洋数据库都表现出较高的覆盖度,可以达到80%以上,而ITS1F相对来说则无法覆盖到足够多的序列。对于反向ITS2来说,允许1个碱基错配的情况下覆盖度会得到大大提升,在Unite数据库和海洋数据库均能覆盖到93%以上的序列。

|
| 图 6 ITS扩增引物对于Unite ITS 3个数据库和海洋真菌ITS 3个数据库的覆盖度 Figure 6 The coverage of ITS amplification primers to 3 Unite databases and 3 marine ITS fungal databases |
|
|
对于ITS全长序列的扩增,收集了3条正向引物ITS1、ITS1F和ITS9MUNngs,以及2条反向引物ITS4R和ITS4ngs。正向引物中,ITS1的覆盖度最高,在允许1个碱基错配的情况下,可以覆盖Unite ITS全长数据库中90%以上的序列,这条正向引物对海洋ITS全长序列数据库也是具有最高的适用性(图 6B)。相对来说,ITS1F和ITS9MUNngs的覆盖度则不够好,当允许1个碱基错配时,ITS1F对于Unite数据库的覆盖度会有一个较大的提升。从引物对来看,ITS1/ITS4R引物对的评价结果最好,能够覆盖到Unite数据库中80%以上的全长序列。然而3对引物对于海洋真菌ITS数据库来说覆盖度都不理想,即使允许了1个碱基错配,覆盖度仍均未达到50%以上。也就是说,这3对引物并不适用于海洋真菌的ITS基因扩增。
针对ITS2区的扩增,收集了4条正向引物gITS7、gITS7ngs、5.8S-Fun和ITS3_KYO2,其中gITS7ngs是在gITS7的基础上增加了2个简并碱基而来(表 4)。4条正向引物对于Unite ITS2数据库的覆盖度都很高,不允许碱基错配的情况下均可达到85%以上,允许一个碱基错配的情况下覆盖度则可以达到90%以上。对于海洋ITS2数据库,这些正向引物的评价结果更好,覆盖度均能达到90%以上。此外还收集了3条反向引物ITS4R、ITS4ngs和ITS4-Fun,其中ITS4ngs是在ITS4R的基础上增加了一个简并碱基,ITS4-Fun则是将ITS4R前端去除第1个碱基,后端延长了6个碱基。从评价结果来看,简并碱基的引入可以在一定程度上提高ITS4R的覆盖度,而引物的增长则会使信息学的评价结果变低,但评价结果差别并不大。从引物对来看,这4对引物的评价结果相差不大,不允许错配的情况下均能覆盖80%以上Unite数据库中的ITS2序列,在允许1个碱基错配的情况下,覆盖度则还能再提升5%左右。此外,这4对引物均能覆盖70%-80%的海洋ITS2序列(图 6C)。
2.3 18S rRNA基因通用引物评价结果通过收集和整理SILVA数据库中18S rRNA基因序列(25 497条),系统地评价了在18S rRNA基因的高通量测序中针对不同区域常用的5对引物,包括4条正向引物和5条反向引物(表 5),其中4对引物是真核微生物通用引物,还有1对引物是针对真菌扩增的特异性引物。因此额外构建了SILVA 18S rRNA基因真菌数据库和非真菌数据库用于评价这些引物对于真菌序列的覆盖度和特异性。此外,基于海洋真菌18S rRNA基因数据库海洋和真菌ITS1序列数据库(表 2)系统评价了18S rRNA基因通用引物对海洋真菌序列的普适性。
由于反向引物EukBr对于真菌序列的覆盖度太低(不足1%),且错配大比例发生在第10个碱基,因此根据BLASTn的比对结果,将第10个碱基T加入碱基C的简并,将原先的引物EukBr (5'-TGAT CCTTCTGCAGGTTCACCTAC-3')改良为EukBr* (5'-TGATCCTTCYGCAGGTTCACCTAC-3')。在不允许错配的情况下,改良后的引物覆盖度可以从原先的不到1%提升至75%以上(图 7)。本文后续对于引物EukBr*的评价均为改良后的结果。此外,由于这条反向引物落在ITS1基因上,因此Unite ITS1基因序列数据库用来评价其覆盖度。

|
| 图 7 18S rRNA基因扩增引物EukBr改进前后对于ITS1数据库的覆盖度 Figure 7 18S rRNA gene primer EukBr's coverage of ITS1 databases before and after the improvement |
|
|
在不允许错配的情况下,7条通用引物中,除了SSU_F_22仅能覆盖55.95%的真核微生物序列,其余6条引物的覆盖度均在70%以上(图 8)。在允许1个碱基错配的情况下,所有引物的覆盖度则均达到了80%以上。由于用来评价反向引物EukBr*数据库与正向引物数据库不同,无法得到扩增V9区的引物对Euk1391f/EukBr*的覆盖度。对于其他4对引物的评价结果来看,扩增V4-V5的引物对TAReuk454FWD1/R1200的覆盖度最高,不允许碱基错配的覆盖度可以达到69.58%,允许1个碱基错配的覆盖度则能达到83.32%。此外,这4对通用引物对真菌序列的适配性也较好,在不允许碱基错配的情况下均能覆盖到60%以上的真菌序列,允许1个碱基错配的覆盖度则都能达到70%以上。用于扩增真菌18S rRNA基因V5区的特异引物对nu-SSU-0817/nu-SSU-1196对于真菌序列的覆盖度反而略低于通用引物。然而,nu-SSU-0817/nu-SSU- 1196对于真菌序列的特异性非常好,在不允许碱基错配的情况下仅能覆盖到0.39%的非真菌序列。对于海洋真菌18S rRNA序列,这5对其他引物的覆盖度都比较高,TAReuk454FWD1/R1200甚至能覆盖到90%以上的海洋真菌序列,SSU_F_22/ SSU_F_9的覆盖度也能达到85%以上(图 8)。

|
| 图 8 18S rRNA基因扩增引物对于对应数据库的覆盖度 Figure 8 Coverage of the 18S rRNA gene amplification primer to their corresponding database |
|
|
下一代测序技术的迅速发展大大降低了微生物多样性分析的检测成本和复杂程度。因此,选择引物扩增标记基因就成为确定序列长度和覆盖范围的关键。2010年,地球微生物组计划(Earth Microbiome Project,EMP)成立,慢慢建立起来自世界各地生态系统的微生物多样性目录[4],以创建微生物组样本数据库。基于此,微生物生态学家可以
控制分析方法的一致性,以促进全球微生物组的比较。因此,EMP提出了标准引物和实验方法,以保证样本间的多样性比较的准确性。对于16S rRNA基因,EMP推荐的标准引物是扩增V4-V5区的515F/806R引物对和扩增V4-V6区的515F/926R引物对。16S rRNA基因的V4-V6区特异性好、数据库信息全,是细菌多样性分析注释的最佳选择。从我们的评价结果来看,在不允许碱基错配的情况
下,515F/806R和515F/926R这2对引物覆盖度也是最高(图 3)。其中,引物对515F/926R的覆盖度略高于515F/806R,但515F/806R的扩增产物长度为291 bp,相对515F/926R的扩增产物长度411 bp更易测序与分析。对于真核生物18S rRNA基因,EMP则推荐了扩增V9区的引物对Euk1391f/EukBr。由于引物EukBr的覆盖度较低,我们对其进行了一个碱基的改良,改良后的引物EukBr*的覆盖度得到了大幅度的提升(图 7)。
对于ITS基因扩增,由于整个ITS区域太长,目前的第二代测序无法对其进行完全测序。因此,我们捕捉的目的片段目前只针对ITS1或ITS2区域,EMP则是推荐了扩增ITS1基因的引物对ITS1F/ITS2。由于这对引物是在真菌物种中只有一小部分分子变异已知的情况下设计的,因此在我们的结果中其覆盖度并不高。在过去的研究中,这对引物在担子菌等部分真菌类群的扩增中有错配,甚至不匹配的比例很高[38, 49],因此当我们允许一个碱基的错配时,这对引物对Unite ITS1数据库的覆盖度可以提高一倍。而这对引物之所以仍然被广泛使用,是因为其对覆盖真菌序列的特异性很好,尤其能避免植物序列的扩增[50]。对于ITS2区域的扩增,EMP并未推荐引物,事实上,ITS1或ITS2究竟哪个片段能更好地反映真菌群落组成仍存有争议。ITS1通常比ITS2短,对物种分辨率低一些,但是扩增和测序错误也低一些;ITS2分辨率高,但是测序的误差会增加OTU的数量。研究表明,在不同的生态环境和群落复杂性下,ITS1和ITS2测序结果反映的群落组成既存在一致性,也存在差异性。例如,在部分土壤和外生菌根真菌群落研究中,ITS1和ITS2呈现出了相似的结果;而在部分土壤和人类肠道真菌群落研究,ITS1和ITS2的结果则存在差异[19]。我们的结果表明,针对ITS2片段的扩增引物覆盖度均高于针对ITS1片段的扩增引物。总之,这些结果表明,真菌群落结构研究所关注的标记基因片段和引物选择尚未标准化。
为了关注真菌的两个标记基因ITS和18S rRNA基因通用引物对于海洋样本的覆盖度,我们还收集了海洋真菌ITS与18S rRNA基因序列构建了海洋数据库。结果表明,覆盖度高的通用引物并不一定能覆盖最多的海洋序列。比如EMP推荐的真核微生物18S rRNA通用引物Euk1391f和EukBr*在不允许错配的情况下分别仅能覆盖60%多的海洋真菌序列,相比其他引物的覆盖度要低一些。对于ITS通用引物来说,扩增ITS2的5.8S-Fun/ITS4-Fun不管对综合数据库还是海洋数据库都表现出较高的覆盖度。我们的结果表明,由于真菌群落结构研究所关注的标记基因片段和引物选择尚未标准化,针对不同的环境样本,标记基因与引物的选择也要另外考量。此外,独特的物种关注偏好可能也需要不同的引物选择、组合或重新设计。
随着我们对未知群落的认识越来越深,信息技术也在不断提升,一些之前设计的引物可以进行改进,以提高对微生物群落认识的准确性。本文扩增ITS2的正向引物gITS7ngs是由gITS7改动了两个碱基,对Unite数据库中真菌ITS序列的覆盖度提升了4.66%,反向引物ITS4ngs则是由ITS4R改动了1个碱基,覆盖度也有所提升,gITS7ngs/ITS4ngs的覆盖度相对于gITS7/ITS4R也提升了5.09%。此外,我们的结果中,许多引物允许1个碱基错配的覆盖度评价结果会比不允许碱基错配时有很大的提升,如ITS2、SSU_F_22等。本文改进了EukBr的第10个碱基,而一些其他引物也还有改进的空间。然而对于引物来说,一个碱基的变化都可能会导致扩增产物发生变化[28]。例如本文中,同一位置的古菌16S rRNA基因引物A806R与通用引物806R相比只有两个碱基的差别,但A806R无法扩增细菌序列。简并碱基的引入也许可以覆盖更多的物种,但也会在一定程度上降低关注物种的特异性。例如,古菌16S rRNA基因通用引物A519F由于一个简并碱基M替换了C,使改良后的A519F*覆盖度提高了2%,但相对地,对细菌序列的覆盖度提升了90%多(图 5)。也就是说,A519F*不再是针对古菌序列具有特异性的古菌引物了。因此,当我们在对引物进行改进时,既要确保能够识别微生物群落及其生存的生态系统之间的联系,还要确保不会引入新的偏差。这除了需要用已知数据库进行覆盖度与特异性的计算,也需要一定的PCR实验与测序数据结果分析。如16S rRNA基因通用引物对515F/806R的简并碱基(Y和N)发生改动时,针对不同环境样本的大量实验用于验证其优势与偏差,结果也表明这些改动适用于海洋、土壤等环境[51]。
在过去的十几年中,下一代测序(NGS)技术被广泛用于探索各种生态系统中微生物群落的多样性和组成结构,对研究海洋、土壤、宿主等环境中的微生物构成有重要的指导作用,同时也是系统发育和分类学研究中一种广泛使用的方法,特别是应用于不同的环境宏基因组学样本中[6]。随着高通量测序技术的不断发展和参考数据库的不断更新,利用核糖体操纵子作为DNA标记可以揭示不同生态系统中的微生物多样性和组成。采用第二代高通量测序平台测定的16S/18S/ITS某个高变区域的序列,可以反映环境样品在细菌、真菌、古菌等分类方面物种之间的差异。然而,针对不同的环境样本,引物的选择和实验过程仍然需要更细致的验证。根据本研究的结果与过去的研究,综合考虑覆盖度、特异性、产物长度的合理性等因素,我们为环境样本的测序引物做了推荐(表 6),为微生态研究中标记基因的引物选择提供了一个广泛的指导,但在关注具体科学问题时,引物的选择仍需数据指导与实验尝试。
| Primers name | EMP proposal | Proposal of this study | Proposal for marine samples |
| 16S rRNA gene universal primers | 515F/806R, 515F/926R | 515F/806R | - |
| 16S rRNA gene archaeal primers | - | A519F/A806R | - |
| ITS1 primers | ITS1F/ITS2 | ITS1F/ITS2, ITS1/ITS2 | ITS1/ITS2 |
| ITS2 primers | - | 5.8S-Fun/ITS4-Fun | 5.8S-Fun/ITS4-Fun |
| 18S rRNA gene primers | Euk1391f/EukBr* | Euk1391f/EukBr* | Euk1391f/EukBr* |
| [1] |
Amann RI. Fluorescently labelled, rRNA-targeted oligonucleotide probes in the study of microbial ecology[J]. Molecular Ecology, 1995, 4(5): 543-554. |
| [2] |
Wei ZY, Jin DC, Deng Y. Bioinformatics tools and applications in the study of environmental microbial metagenomics[J]. Microbiology China, 2015, 42(5): 890-901. (in Chinese) 魏子艳, 金德才, 邓晔. 环境微生物宏基因组学研究中的生物信息学方法[J]. 微生物学通报, 2015, 42(5): 890-901. |
| [3] |
Zhang JY, Zhu BC, Xu C, et al. Strategy of selecting 16S rRNA hypervariable regions for matagenome-phylogenetic marker genes based analysis[J]. Chinese Journal of Applied Ecology, 2015, 26(11): 3545-3553. (in Chinese) 张军毅, 朱冰川, 徐超, 等. 基于分子标记的宏基因组16S rRNA基因高变区选择策略[J]. 应用生态学报, 2015, 26(11): 3545-3553. |
| [4] |
Gilbert JA, Meyer F, Jansson J, et al. The Earth Microbiome Project: Meeting report of the "1st EMP meeting on sample selection and acquisition" at Argonne National Laboratory October 6th 2010[J]. Standards in Genomic Sciences, 2010, 3(3): 249-253. DOI:10.4056/aigs.1443528 |
| [5] |
Tedersoo L, Anslan S, Bahram M, et al. Shotgun metagenomes and multiple primer pair-barcode combinations of amplicons reveal biases in metabarcoding analyses of fungi[J]. MycoKeys, 2015, 10: 1-43. DOI:10.3897/mycokeys.10.4852 |
| [6] |
Gohl DM, Vangay P, Garbe J, et al. Systematic improvement of amplicon marker gene methods for increased accuracy in microbiome studies[J]. Nature Biotechnology, 2016, 34(9): 942-949. DOI:10.1038/nbt.3601 |
| [7] |
Zhou JZ, Xue K, Xie JP, et al. Microbial mediation of carbon-cycle feedbacks to climate warming[J]. Nature Climate Change, 2012, 2(2): 106-110. |
| [8] |
Du XF, Yu H, Wang S, et al. Metagenomics reveal responses of soil microbial community in grassland to global changes[J]. Chinese Journal of Ecology, 2019, 38(11): 3516-3526. (in Chinese) 杜雄峰, 于皓, 王尚, 等. 宏基因组方法揭示草地土壤微生物群落响应全球变化[J]. 生态学杂志, 2019, 38(11): 3516-3526. |
| [9] |
Wang ZJ, Lu GX, Yuan MT, et al. Elevated temperature overrides the effects of N amendment in Tibetan grassland on soil microbiome[J]. Soil Biology and Biochemistry, 2019, 136: 107532. DOI:10.1016/j.soilbio.2019.107532 |
| [10] |
Wu LW, Ning DL, Zhang B, et al. Global diversity and biogeography of bacterial communities in wastewater treatment plants[J]. Nature Microbiology, 2019, 4(7): 1183-1195. DOI:10.1038/s41564-019-0426-5 |
| [11] |
Maron PA, Mougel C, Ranjard L. Soil microbial diversity: methodological strategy, spatial overview and functional interest[J]. Comptes Rendus Biologies, 2011, 334(5/6): 403-411. |
| [12] |
Simon HM, Smith MW, Herfort L. Metagenomic insights into particles and their associated microbiota in a coastal margin ecosystem[J]. Frontiers in Microbiology, 2014, 5: 466. |
| [13] |
Wlasiuk G, Vercelli D. The farm effect, or: when, what and how a farming environment protects from asthma and allergic disease[J]. Current Opinion in Allergy and Clinical Immunology, 2012, 12(5): 461-466. DOI:10.1097/ACI.0b013e328357a3bc |
| [14] |
Li SZ, Deng Y, Zhang ZJ, et al. Advances in molecular detection on aromatic bioremediation[J]. China Environmental Science, 2019, 39(6): 2577-2587. (in Chinese) 厉舒祯, 邓晔, 张照婧, 等. 生物降解芳香族化合物的分子检测技术研究进展[J]. 中国环境科学, 2019, 39(6): 2577-2587. |
| [15] |
Wu YN, Yang YZ, Cao L, et al. Habitat environments impacted the gut microbiome of long-distance migratory swan geese but central species conserved[J]. Scientific Reports, 2018, 8(1): 13314. DOI:10.1038/s41598-018-31731-9 |
| [16] |
Reyes A, Semenkovich NP, Whiteson K, et al. Going viral: next-generation sequencing applied to phage populations in the human gut[J]. Nature Reviews Microbiology, 2012, 10(9): 607-617. DOI:10.1038/nrmicro2853 |
| [17] |
Simon C, Daniel R. Metagenomic analyses: past and future trends[J]. Applied and Environmental Microbiology, 2011, 77(4): 1153-1161. |
| [18] |
Frank JA, Reich CI, Sharma S, et al. Critical evaluation of two primers commonly used for amplification of bacterial 16S rRNA genes[J]. Applied and Environmental Microbiology, 2008, 74(8): 2461-2470. DOI:10.1128/AEM.02272-07 |
| [19] |
Li S, Deng Y, Wang Z, et al. Exploring the accuracy of amplicon-based internal transcribed spacer markers for a fungal community[J]. Molecular Ecology Resources, 2020, 20(1): 170-184. DOI:10.1111/1755-0998.13097 |
| [20] |
Bradley IM, Pinto AJ, Guest JS. Design and evaluation of Illumina MiSeq-compatible, 18S rRNA gene-specific primers for improved characterization of mixed phototrophic communities[J]. Applied and Environmental Microbiology, 2016, 82(19): 5878-5891. DOI:10.1128/AEM.01630-16 |
| [21] |
Lane DJ. 16S/23S rRNA sequencing[A]//Stackebrandt E, Goodfellow M. Nucleic Acid Techniques in Bacterial Systematics[M]. New York: John Wiley and Sons, 1991: 115-175
|
| [22] |
Winsley T, van Dorst JM, Brown MV, et al. Capturing greater 16S rRNA gene sequence diversity within the domain bacteria[J]. Applied and Environmental Microbiology, 2012, 78(16): 5938-5941. DOI:10.1128/AEM.01299-12 |
| [23] |
Caporaso JG, Lauber CL, Walters WA, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample[J]. Proceedings of the National Academy of Sciences of the United States of America, 2011, 108(Supplement 1): 4516-4522. |
| [24] |
Brosius J, Palmer ML, Kennedy PJ, et al. Complete nucleotide sequence of a 16S ribosomal RNA gene from Escherichia coli[J]. Proceedings of the National Academy of Sciences of the United States of America, 1978, 75(10): 4801-4805. DOI:10.1073/pnas.75.10.4801 |
| [25] |
Vescio PA, Nierzwicki-Bauer SA. Extraction and purification of PCR amplifiable DNA from lacustrine subsurface sediments[J]. Journal of Microbiological Methods, 1995, 21(3): 225-233. DOI:10.1016/0167-7012(94)00044-8 |
| [26] |
Turner S, Pryer KM, Miao VPW, et al. Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRNA sequence analysis[J]. The Journal of Eukaryotic Microbiology, 1999, 46(4): 327-338. DOI:10.1111/j.1550-7408.1999.tb04612.x |
| [27] |
Caporaso JG, Lauber CL, Walters WA, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms[J]. The ISME Journal, 2012, 6(8): 1621-1624. DOI:10.1038/ismej.2012.8 |
| [28] |
Parada AE, Needham DM, Fuhrman JA. Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples[J]. Environmental Microbiology, 2016, 18(5): 1403-1414. DOI:10.1111/1462-2920.13023 |
| [29] |
Lane DJ, Pace B, Olsen GJ, et al. Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses[J]. Proceedings of the National Academy of Sciences of the United States of America, 1985, 82(20): 6955-6959. DOI:10.1073/pnas.82.20.6955 |
| [30] |
Macrae A. The use of 16S rDNA methods in soil microbial ecology[J]. Brazilian Journal of Microbiology, 2000, 31(2): 77-82. |
| [31] |
Stahl D, Amann R. Development and application of nucleic acid probes[A]//Stackebrandt E, Goodfellow M. Nucleic Acid Techniques in Bacterial Systematics[M]. Chichester: John Wiley & Sons Ltd., 1991: 205-248
|
| [32] |
Ovreås L, Forney L, Daae FL, et al. Distribution of bacterioplankton in meromictic Lake Saelenvannet, as determined by denaturing gradient gel electrophoresis of PCR-amplified gene fragments coding for 16S rRNA[J]. Applied and Environmental Microbiology, 1997, 63(9): 3367-3373. DOI:10.1128/AEM.63.9.3367-3373.1997 |
| [33] |
Wang N, Chang ZZ, Xue XM, et al. Biochar decreases nitrogen oxide and enhances methane emissions via altering microbial community composition of anaerobic paddy soil[J]. Science of the Total Environment, 2017, 581-582: 689-696. DOI:10.1016/j.scitotenv.2016.12.181 |
| [34] |
Wang YZ, Feng XY, Natarajan VP, et al. Diverse anaerobic methane-and multi-carbon alkane-metabolizing archaea coexist and show activity in Guaymas Basin hydrothermal sediment[J]. Environmental Microbiology, 2019, 21(4): 1344-1355. DOI:10.1111/1462-2920.14568 |
| [35] |
White TJ, Bruns TD, Lee SB, et al. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics[A]//PCR Protocols: A Guide to Methods and Applications[M]. San Diego: Academic Press, 1990: 315-322
|
| [36] |
Gardes M, Bruns TD. ITS primers with enhanced specificity for basidiomycetes-application to the identification of mycorrhizae and rusts[J]. Molecular Ecology, 1993, 2(2): 113-118. |
| [37] |
Ihrmark K, Bödeker ITM, Cruz-Martinez K, et al. New primers to amplify the fungal ITS2 region–evaluation by 454-sequencing of artificial and natural communities[J]. FEMS Microbiology Ecology, 2012, 82(3): 666-677. DOI:10.1111/j.1574-6941.2012.01437.x |
| [38] |
Tedersoo L, Lindahl B. Fungal identification biases in microbiome projects[J]. Environmental Microbiology Reports, 2016, 8(5): 774-779. DOI:10.1111/1758-2229.12438 |
| [39] |
Taylor DL, Walters WA, Lennon NJ, et al. Accurate estimation of fungal diversity and abundance through improved lineage-specific primers optimized for Illumina amplicon sequencing[J]. Applied and Environmental Microbiology, 2016, 82(24): 7217-7226. DOI:10.1128/AEM.02576-16 |
| [40] |
Toju H, Tanabe AS, Yamamoto S, et al. High-coverage ITS primers for the DNA-based identification of ascomycetes and basidiomycetes in environmental samples[J]. PLoS One, 2012, 7(7): e40863. DOI:10.1371/journal.pone.0040863 |
| [41] |
Tedersoo L, Anslan S. Towards PacBio-based pan-eukaryote metabarcoding using full-length ITS sequences[J]. Environmental Microbiology Reports, 2019, 11(5): 659-668. DOI:10.1111/1758-2229.12776 |
| [42] |
Tedersoo L, Bahram M, Põlme S, et al. Global diversity and geography of soil fungi[J]. Science, 2014, 346(6213): 1256688. DOI:10.1126/science.1256688 |
| [43] |
Bengtsson-Palme J, Ryberg M, Hartmann M, et al. Improved software detection and extraction of ITS1 and ITS2 from ribosomal ITS sequences of fungi and other eukaryotes for analysis of environmental sequencing data[J]. Methods in Ecology and Evolution, 2013, 4(10): 914-919. |
| [44] |
Blaxter ML, de Ley P, Garey JR, et al. A molecular evolutionary framework for the phylum Nematoda[J]. Nature, 1998, 392(6671): 71-75. DOI:10.1038/32160 |
| [45] |
Stoeck T, Bass D, Nebel M, et al. Multiple marker parallel tag environmental DNA sequencing reveals a highly complex eukaryotic community in marine anoxic water[J]. Molecular Ecology, 2010, 19(2): 21-31. |
| [46] |
Borneman J, Hartin RJ. PCR primers that amplify fungal rRNA genes from environmental samples[J]. Applied and Environmental Microbiology, 2000, 66(10): 4356-4360. DOI:10.1128/AEM.66.10.4356-4360.2000 |
| [47] |
Medlin L, Elwood HJ, Stickel S, et al. The characterization of enzymatically amplified eukaryotic 16S-like rRNA-coding regions[J]. Gene, 1988, 71(2): 491-499. |
| [48] |
Hadziavdic K, Lekang K, Lanzen A, et al. Characterization of the 18S rRNA gene for designing universal eukaryote specific primers[J]. PLoS One, 2014, 9(2): e87624. DOI:10.1371/journal.pone.0087624 |
| [49] |
Bellemain E, Carlsen T, Brochmann C, et al. ITS as an environmental DNA barcode for fungi: an in silico approach reveals potential PCR biases[J]. BMC Microbiology, 2010, 10(1): 189. |
| [50] |
Leff JW, Bardgett RD, Wilkinson A, et al. Predicting the structure of soil communities from plant community taxonomy, phylogeny, and traits[J]. The ISME Journal, 2018, 12: 1794-1805. DOI:10.1038/s41396-018-0089-x |
| [51] |
Walters W, Hyde ER, Berg-Lyons D, et al. Improved bacterial 16S rRNA gene (V4 and V4-5) and fungal internal transcribed spacer marker gene primers for microbial community surveys[J]. mSystems, 2016, 1(1): e00009-15. |