 2020, Vol. 47
2020, Vol. 47扩展功能
文章信息
- 张景欣, 沈会芳, 蒲小明, 孙大元, 杨祁云, 林壁润
- ZHANG Jing-Xin, SHEN Hui-Fang, PU Xiao-Ming, SUN Da-Yuan, YANG Qi-Yun, LIN Bi-Run
- 引起芭蕉芋细菌性软腐病的玉米迪基氏菌(Dickeya zeae)CE1的全基因组序列及比较基因组学分析
- Comparative genomics analysis of Dickeya zeae CE1 causing bacterial soft rot of Canna edulis
- 微生物学通报, 2020, 47(11): 3600-3613
- Microbiology China, 2020, 47(11): 3600-3613
- DOI: 10.13344/j.microbiol.china.200001
-
文章历史
- 收稿日期: 2020-01-01
- 接受日期: 2020-02-20
- 网络首发日期: 2020-03-19





迪基氏菌属(Dickeya genus)是由菊欧文氏菌(Erwinia chrysanthemi)建立的一个新属,原隶属于肠杆菌科(Enterobacteriaceae),现与果胶杆菌属(Pectobacterium)隶属于一个新科,即果胶杆菌科(Pectobacteriaceae)[1]。Dickeya属细菌可在全世界范围内对多种重要作物产生严重危害,包括水稻、香蕉、玉米、兰花属、菊花属、石竹类属、马铃薯、番茄、梨树等[2]。据报道,玉米迪基氏菌(Dickeya zeae)是该属中危害作物较多的一个种,如对香蕉、水稻、芭蕉芋等重要作物的危害,其中多个研究[3-5]为本研究团队在国内的首次报道。
芭蕉芋(Canna edulis Ker.),又名旱藕、蕉藕、姜芋,分类上属美人蕉科美人蕉属,为多年生草本植物,具块状根茎。芭蕉芋具有适应性广、产量高、易栽培、抗性强和用途广等特点[6],自20世纪初引入我国栽培后,在云南、贵州、四川、广东和广西等亚热带省区广泛种植[7]。由于块茎淀粉含量高达66%以上,芭蕉芋成为粮食、能源、饲料等多用途兼用的经济作物[6];随着深层次提取与加工技术的发展,芭蕉芋也可作为高价值淀粉、酿造发酵、食品和饲料加工、医药工业的原料进行开发应用。单以种植环节的鲜块茎收购为例,块茎一般产量在45.0−52.5 t/hm2,鲜块茎单价一般为700元/t,产值在31 500−33 750元/hm2,具有较高的经济价值,这使得芭蕉芋产业也具备一定的规模,如贵州现有芭蕉芋种植面积为4万hm2[8]。现阶段,芭蕉芋病虫害发生较少且相关的报道也少[9],有报道零星芭蕉芋植株茎叶出现水渍状病斑,但是其病因和病理不清楚,也难以防治[10]。2017年7月,本研究团队在广东省梅州市五华县发现多个芭蕉芋的种植田发生严重的细菌性病害,约20%的芭蕉芋产生危害症状,经分离鉴定,该细菌性病害是由D. zeae CE1菌株侵染引起的细菌性软腐病;该报道是在国内首次发现由D. zeae引起的芭蕉芋细菌性软腐病,具体发病症状包括:茎基部变黑,出现水渍状坏死病斑;病斑快速扩张,茎基部组织变软、腐烂,最终整株死亡[5]。迄今尚未见有关于芭蕉芋细菌性软腐病的其他相关报道,也很少见报道芭蕉芋发生其他的病虫害,这个可能与芭蕉芋对于生物或者非生物因素的抗性较强有关系[6]。然而芭蕉芋一旦受到D. zeae CE1病原菌的侵染,会产生毁灭性的危害。
除D. zeae NCPPB 3531和NCPPB 3532分离于澳大利亚的马铃薯及D. zeae CSL RW192和MK19分离于水体外[11],D. zeae菌株现阶段报道的菌株多是侵染单子叶植物,如D. zeae Ech586分离于美国佛罗里达的喜林芋,MS1和MS2分离于香蕉,EC1、EC2、ZJU1202和DZ2Q分离于水稻,CE1则分离于芭蕉芋;其中,CE1、MS1、MS2、EC1、EC2和ZJU1202病原菌株全部来源于广东。芭蕉芋分类上属于芭蕉目(Scitamineae)美人蕉科(Cannaceae)美人蕉属(Canna),香蕉分类上属于芭蕉目(Scitamineae)芭蕉科(Musaceae)芭蕉属(Musa),它们在分类上分别属芭蕉目的两个科,亲缘关系较为接近。那么,芭蕉芋菌株CE1与同样来源于广东的香蕉菌株MS1、MS2之间的遗传亲缘关系如何,与来源于广东水稻菌株的遗传分化特征又是如何,这些至今都尚属未知。因此,本研究对芭蕉芋细菌性软腐病菌D. zeae CE1进行全基因组测序和分析,利用以上6个来源于广东的芭蕉芋、香蕉和水稻的D. zeae菌株进行了比较基因组学分析,以揭示D. zeae种内不同菌株在与寄主互作过程中可能存在的遗传分化关系,这对于研究D. zeae种内菌株遗传分化、与寄主互作相关的致病机制等具有重要意义。
1 材料与方法 1.1 细菌菌株芭蕉芋细菌性软腐病菌株,D. zeae CE1由本研究团队分离鉴定[5],菌种保存于广东省植物保护新技术重点实验室,在−80 ℃下保存于甘油备用。
1.2 主要试剂和仪器及培养基细菌基因组提取试剂盒,天根生化科技(北京)有限公司。Pacific Biosciences RSII测序仪,Pacific Biosciences公司;Illumina HiSeq 4000,Illumina公司。
LB液体培养基(g/L):细菌性蛋白胨10.0,酵母提取粉5.0,NaCl 10.0,pH 7.0。固体培养基加1.0 g/L琼脂粉。
1.3 细菌基因组DNA的提取挑取−80 ℃保存的D. zeae CE1菌液在LB固体培养基上划线,32 ℃静置培养24 h。挑取单菌落于LB液体培养基中,32 ℃、150 r/min培养过夜,培养至108 cfu/mL。取4 mL的细菌培养物,使用细菌基因组提取试剂盒提取DNA。
1.4 细菌基因组的三代全基因组测序和二代测序校正经Qubit和NanoDrop检测提取的D. zeae CE1基因组DNA合格后,将该基因组DNA片段化,再使用G-tubes (Covaris)末端处理DNA,下一步用于构建SMRTbell DNA文库(> 10 kb);SMRT文库由广州基迪奥生物科技有限公司在Pacific Biosciences RSII测序仪上进行三代测序,使用广州基迪奥生物科技有限公司自编的数据过滤脚本SMRTanalysis (v2.3.0)对FASTQ测序原始数据进行过滤,选择≥100 bp且质量值在0.80以上的序列用于下一步分析。
分层基因组组装流程(hierarchical genome- assembly process pipeline)[12]通过将短序列比对到长的核心序列(≥6 kb)上以用于修正测序随机错误,再用Celera组装方法的序列重叠区域-布局图-一致性序列(overlap-layout-consensus,OLC)策略进行de novo组装[13],然后用Quiver算法对组装的基因组序列进行质量验证[12]。剔除组装的基因组序列的末端,拼接获得环状DNA结构的基因组序列。构建双末端DNA (paired-end DNA)文库,并送往广州基迪奥生物科技有限公司用Illumina HiSeq 4000进行二代测序,用Burrows-Wheeler比对工具[14]将二代测序序列比对于三代测序组装完成的环状DNA基因组序列上,用Pilon工具[15]查找三代测序序列和二代测序序列间的差异并修正,从而获得完整的环状DNA结构的基因组序列。以上基因组组装、序列差异比对及校正等软件的使用均按照默认参数。
1.5 基因组注释将获得的细菌完整基因组序列使用RepeatMasker[16]、rRNAmmer[17]、tRNAscan[18]和GeneMarkS[19]分别进行重复序列、非编码RNA、tRNA和编码基因的预测;使用Circos[20]将细菌基因组的(G+C)mol%含量、GC偏移值(GC skew,[G−C]/[G+C])、tRNA、rRNA和正义链或负义链上的预测基因分别展示到基因组圈图上。预测的编码基因使用NCBI Non-redundant Protein、UniProt/Swiss-Prot、Kyoto Encyclopedia of Genes and Genomes、Gene Ontology、Clusters of Orthologous Groups和Protein Families数据库进行比对,注释基因功能;同时用CRISPRFinder[21]和antiSMASH 4.0[22]分别对CRISPR序列和代谢产物合成相关基因簇进行预测。以上基因组注释软件的使用均按照默认参数。
1.6 Dickeya zeae种内菌株的基因组比较分析选择NCBI数据库中13个具有全基因组序列的D. zeae菌株作进化树分析,并以D. dadantii 3937菌株作为种外对照菌株。提取以上菌株的dnaX、recA、dnaN、fusA、gapA、purA、rplB、rpoS和gyrA等9个基因的序列,按顺序连接后用于进化树分析。使用MEGA 5.1软件作进化树分析,分析方法采用邻接法(neighbor-joining method)和1 000个重复(replications)的bootstrap值。用CLC Genomics Workbench 12.0.3对不同D. zeae菌株间的平均核苷酸一致性(average nucleotide identity,ANI)值和比对序列百分比(alignment percentage,AP)值进行估算,同时使用Genome-to-Genome Distance Calculator 2.1估算菌株间的电子DNA-DNA杂交(in silico DNA-DNA hybridization,isDDH)值;用Mummer软件进行菌株间的共线性分析。用OrthoMCL分析方法[23]进行D. zeae菌株的基因家族分析,在BLAST比对中选择的p值的阈值为1×10−5,相似性的阈值为80%。
2 结果与分析 2.1 Dickeya zeae CE1的全基因组序列组装及基本信息三代测序产生的原始数据经过滤后获得76 899条≥100 bp且质量值在0.80以上的序列,平均长度在11 859 bp,共计911 997 636 bp;de novo组装获得4 713 002 bp的基因组序列,经过滤后的测序序列的基因组覆盖倍数达193倍。二代测序产生17 940 732条序列,其中17 703 508条(占98.68%)序列可比对上三代测序的组装序列;经过比对,增加了1 734 bp的点序列和删减了5 bp的点序列,最终获得4 714 731 bp的完整基因组序列,其(G+C)mol%含量为53.63%;该基因组组装程度达到单一长序列片段(scaffold)的水平,基因组内无gap。基因组注释预测到4 052个编码基因、75个tRNA、9个ncRNA及7个rRNA (图 1)。该完整基因组的序列及注释信息已上传至NCBI GenBank,登录号为CP033622。

|
| 图 1 D. zeae CE1细菌完整基因组圈图 Figure 1 Circular visualization of the complete genome of D. zeae CE1 |
|
|
通过基于9个保守基因串联序列的进化树分析发现,13个属于D. zeae种的菌株和1个种外的对照菌株D. dadantii 3937可分成明显的两类;而D. zeae种的菌株可分为三小类(图 2),分别为:(1)水稻菌株EC1、ZJU1202、DZ2Q和EC2,以及其他菌株如CSL RW192、NCPPB 3531和Ech586;(2)芭蕉芋菌株CE1和香蕉菌株MS1、MS2,以及其他菌株如NCPPB 3532和MK19;(3) NCPPB 2538。其中,水稻菌株EC1、ZJU1202、EC2和芭蕉芋菌株CE1及香蕉菌株MS1、MS2均是分离于广东的病原菌。芭蕉芋菌株CE1和其他D. zeae菌株的ANI、AP和isDDH值与进化树分析反映的菌株间亲缘关系类似,CE1和香蕉菌株MS1、MS2以及其他菌株NCPPB 3532、MK19间的ANI、AP、isDDH值相较于CE1和以水稻菌株为主的第一类菌株的值要大(表 1)。

|
| 图 2 D. zeae菌株基于9个保守基因串联序列的进化树分析 Figure 2 Phylogenetic tree of D. zeae strains based on concatenated sequences of 9 genes 注:菌株名称后括号内序列号代表该菌株基因组序列的NCBI GenBank登录号;进化树分支点的数字代表该分支点的bootstrap值,数值以百分比(%)形式展示;标尺数字代表该长度的分支所代表的基因组遗传变异度. Note:The numbers within brackets following the bacterial strains indicate the NCBI GenBank accession numbers for their genome sequences; The numbers at the nodes of different branches in the phylogenetic tree indicate the bootstrap values, which are displayed in the form of percentage (%); The number of scale bar indicates the genetic variability of the genome sequences represented by the branch of this determined length. |
|
|
| Strains compared to strain CE1 | ANI | AP | isDDH |
| MS1 | 96.05 | 85.4 | 67.3 |
| MS2 | 96.17 | 86.19 | 68.3 |
| NCPPB_3532 | 96.2 | 87.4 | 68.2 |
| MK19 | 96.16 | 86.61 | 68.0 |
| EC2 | 94.3 | 82.67 | 57.1 |
| EC1 | 94.13 | 79.51 | 56.2 |
| ZJU 1202 | 94.11 | 79.74 | 56.0 |
| DZ 2Q | 94.12 | 79.27 | 56.0 |
| NCPPB_3531 | 94.52 | 84.48 | 57.8 |
| CSL_RW192 | 94.41 | 81.84 | 57.5 |
| Ech 586 | 95.69 | 85.33 | 64.9 |
| NCPPB_2538 | 96.14 | 87.69 | 68.4 |
D. zeae菌株基因家族分析发现,3 146个基因家族是6个广东菌株共有的,包括CE1菌株的3 211个基因、MS1菌株的3 207个基因、MS2菌株的3 213个基因、EC1菌株的3 206个基因、ZJU1202菌株的3 195个基因以及EC2菌株的3 215个基因,共计19 247个保守基因,见图 3。以上结果表明,多数基因在6个菌株间均是保守的,仅小部分基因是各个菌株所特有的。其中,水稻菌株EC1、ZJU1202所特有的基因最少,另一个水稻菌株EC2次之,表明了水稻菌株间遗传分化程度较低;这与9个用于进化树分析的保守基因串联序列的核苷酸多态性分析一致,3个水稻菌株的核苷酸多态性(Pi)为0.009 25,低于芭蕉芋和香蕉菌株间的核苷酸多态性0.019 77。相较之下,芭蕉芋菌株CE1和香蕉菌株MS1、MS2各自所特有的基因则相对较多,特别是芭蕉芋菌株,包含260个基因的260个基因家族是芭蕉芋菌株所特有的。

|
| 图 3 6个广东D. zeae菌株基因家族分析的维恩图 Figure 3 Venn diagram of common or specific genes among 6 Guangdong D. zeae strains by OrthoMCL analysis 注:上方的数字代表基因家族的数量,下方在括号内的数字代表基因数量. Note: Upper numbers indicate the number of groups of genes, lower numbers in parentheses indicate the number of genes. |
|
|
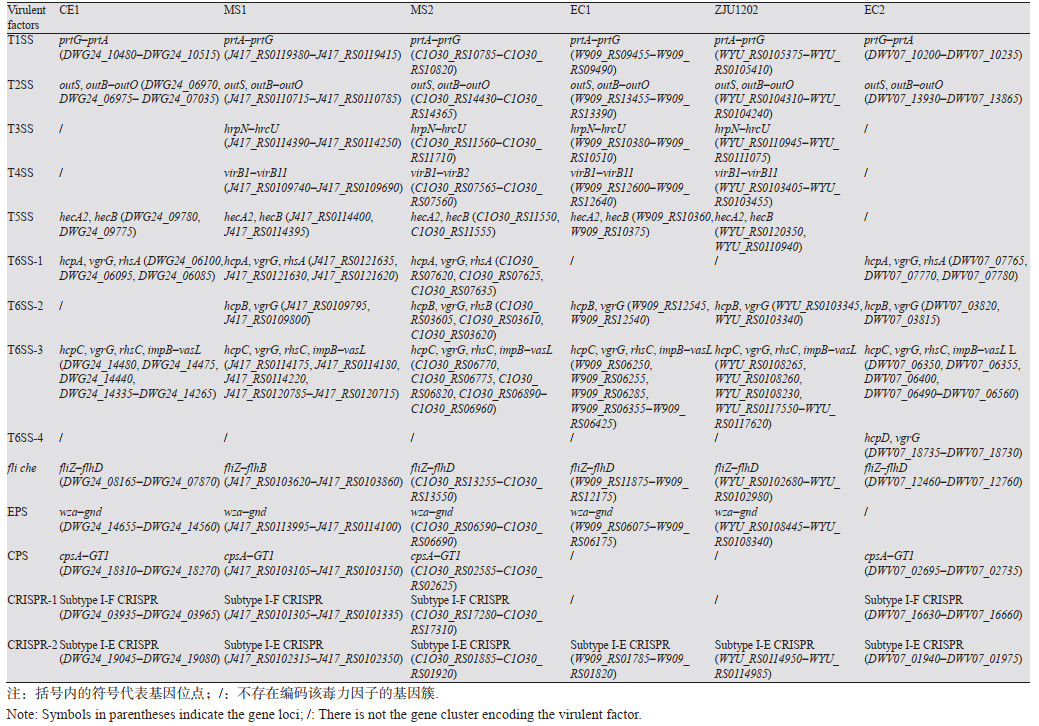
通过功能注释,在各菌株所特异的基因中并未发现与细菌分泌系统、鞭毛、胞外多糖、CRISPR等重要致病因子相关的基因或者基因簇。分析编码细菌分泌系统、鞭毛蛋白、胞外多糖、CRISPR等基因或基因簇在6个广东菌株间的保守情况(表 2)发现,细菌分泌系统中仅Ⅰ型(T1SS)、Ⅱ型(T2SS)和Ⅵ型分泌系统(T6SS)在6个菌株中全部存在。在T6SS中,T6SS-3位点具有核心基因簇impB−vasL,而且只有T6SS-3位点在6个菌株中均存在;水稻菌株EC1、ZJU1202缺失T6SS-1而芭蕉芋菌株CE1缺失T6SS-2位点,T6SS-4位点仅存在于水稻菌株EC2。Ⅲ型(T3SS)、Ⅳ型(T4SS)、Ⅴ型分泌系统(T5SS)在水稻菌株EC2中是全部缺失的;芭蕉芋菌株缺失了T3SS和T4SS,而香蕉菌株MS2在T4SS上仅具有virB1−virB2基因而缺失其余大部分基因。鞭毛基因簇fliZ−flhD在6个菌株间均是保守的,仅是MS1在cheZ−flhD区域被转座酶等基因替代。胞外多糖合成基因簇EPS和CPS基因簇在芭蕉芋和香蕉菌株中是保守的;但是,水稻菌株在该位点上表现明显的分化,菌株EC1和ZJU1202仅具有EPS基因簇,菌株EC2则仅发现CPS基因簇。两类CRISPR序列subtype I-F CRISPR、Subtype I-E CRISPR在芭蕉芋、香蕉菌株中同样是保守的;在水稻菌株中,仅EC2具有两类CRISPR序列,而EC1和ZJU1202则缺失了Subtype I-F CRISPR。以上结果表明,部分基因组区域如T1SS、T2SS、T6SS-3、鞭毛基因簇在两类寄主来源的菌株间保守,部分区域如T3SS、T4SS、T5SS在不同菌株间均表现较高程度的分化,但是未表现与寄主相关的分化;另外,EPS、CPS和CRISPR等区域表现一定程度与寄主相关的分化,如它们在芭蕉芋和香蕉菌株间是保守的,但是在水稻菌株内表现明显分化。因此,细菌分泌系统、鞭毛、胞外多糖、CRISPR等重要致病因子在两类不同寄主来源的菌株间并未表现明显的遗传分化;下一步拟对两类不同寄主来源的菌株所特异的基因进行功能预测分析。
|
6个来源于广东不同类型寄主的菌株基因家族分析表明,80个基因是芭蕉芋菌株CE1及香蕉菌株MS1、MS2所特有的,42个基因则是水稻菌株EC1、ZJU1202和EC2所特有的。
芭蕉芋菌株CE1及香蕉菌株MS1、MS2特异基因的COG功能分析表明,分别有19、14、12和9个特异基因的COG功能预测为一般功能(general function prediction)、氨基酸转运和代谢(amino acid transport and metabolism)、转录(transcription)及脂质转运和代谢(lipid transport and metabolism) (图 4)。进一步比对发现,有2个基因簇(图 5)的基因较多属于以上4类COG功能。第1个基因簇包括vdlC、fabF、SPCC162.03 3个基因(图 5A),KEGG通路分析表明,fabF基因参与了代谢过程(metabolic pathways)、脂肪酸合成(fatty acid biosynthesis)、生物素代谢(biotin metabolism)、脂肪酸代谢(fatty acid metabolism)等代谢通路;经寄主-病原物互作数据库(PHI-base)分析,该基因簇上3个基因均可比对上已知的与病原菌-植物互作相关的功能基因,且主要是与单子叶植物互作病原菌的功能基因,如fabF比对上的基因编码了聚酮合酶(polyketide synthase,PKs)。第2个基因簇包括gsiB、NGR_a01400、NGR_a01410、appC、gsiC、vanR、laaA 7个基因(图 5B),KEGG通路分析发现,该基因簇的6个基因参与了群体感应(quorum sensing)的代谢通路,其中vanR同时参与了双元系统(two-component system)和生物被膜形成(biofilm formation)等代谢通路;PHI-base分析表明,NGR_a01400、gsiC、vanR可比对上已知的与病原菌-植物互作相关的功能基因,这些基因同样主要是与单子叶植物互作的病原菌的功能基因,其中值得注意的是vanR可比对上引起水稻穗枯病的水稻细菌性谷枯病菌(Burkholderia glumae)的tofR基因,该基因与毒黄素(toxoflavin)合成相关。

|
| 图 4 芭蕉芋和香蕉菌株特异基因的COG功能注释 Figure 4 COG function annotation of specific genes within C. edulis and banana D. zeae strains |
|
|

|
| 图 5 芭蕉芋和香蕉菌株2个具代表性的特异基因簇 Figure 5 Two representative specific gene clusters within C. edulis and banana D. zeae strains 注:A:在芭蕉芋和香蕉菌株中特异的vdlC–SPCC162.03基因簇,B:在芭蕉芋和香蕉菌株中特异的gsiB–laaA基因簇.上方的符号代表基因名称,下方的符号代表各个基因分别在D. zeae菌株CE1、MS1和MS2的基因位点;DWG24代表菌株CE1,J417代表菌株MS1,C1030代表菌株MS2. Note: A: gene cluster of vdlC–SPCC162.03 specific to C. edulis and banana strains; B: gene cluster of gsiB–laaA specific to C. edulis and banana strains. the symbols above indicate the gene symbols, and the symbols below indicate the gene loci within D. zeae CE1, MS1 and MS2; DWG24 indicates strain CE1, J417 indicates strain MS1, and C1030 indicates strain MS2. |
|
|
水稻菌株EC1、ZJU1202和EC2特异基因的COG功能分析表明,分别有15、7和5个特异基因的COG功能预测为碳水化合物转运和代谢(carbohydrate transport and metabolism)、转录及一般功能(图 6)。在预测为碳水化合物转运和代谢的基因中发现有两个基因簇,其中较大的一个基因簇包括yulB、rbsA2、rbsC、lsrD、alsE、alsK、rbsK等7个基因(图 7A),KEGG通路分析表明,alsE、alsK同时参与了多种环境下的微生物代谢(microbial metabolism in diverse environments)及果糖和甘露糖代谢(fructose and mannose metabolism)两个代谢通路;经PHI-base分析,rbsA2可比对上稻瘟病菌的ABC4蛋白,该蛋白与多重耐药性(multidrug resistance)相关;碳水化合物活性酶数据库(CAZy)分析发现,rbsK可比对上多个糖苷水解酶(glycoside hydrolase,GH)蛋白。另外,在Type I-E CRISPR位点的上游邻接基因中发现了一个基因簇是水稻菌株所特有的(图 7B),该基因簇的3个基因分别编码MFS转运蛋白(MFS transporter)、dTDP-6-脱氧-D-葡萄糖-3, 5-表异构酶(dTDP- 6-deoxy-D-glucose-3, 5-epimerase)和XRE家族转录调控因子(XRE family transcriptional regulator)。

|
| 图 6 水稻菌株特异基因的COG功能注释 Figure 6 COG function annotation of specific genes within rice D. zeae strains |
|
|

|
| 图 7 水稻菌株的2个具代表性的特异基因簇 Figure 7 Two representative specific gene clusters within rice D. zeae strains 注:A:在水稻菌株中特异的yulB–rbsK基因簇;B:在Type I-E CRISPR上游邻接位点发现的水稻菌株特异基因簇.上方的符号代表基因名称,下方的符号代表各个基因分别在D. zeae菌株EC1、EC2和ZJU1202的基因位点;W909代表菌株EC1,WYU代表菌株ZJU1202,DWV07代表菌株EC2. Note: A: gene cluster of yulB–rbsK specific to rice strains; B: the specific gene cluster from rice strains found in the adjacent genomic locus of upstream of the type I-E CRISPR array. the symbols above indicate the gene symbols, and the symbols below indicate the gene loci within D. zeae EC1, EC2 and ZJU1202. W909 indicates strains EC1, WYU indicates strains ZJU1202, and DWV07 indicates strains EC2. |
|
|
Dickeya zeae菌株在遗传基础和表型性状等方面的差异较大[24-25],本研究中D. zeae种内菌株的进化树分析也表明,这些菌株具有较大程度的遗传分化(图 2)。D. zeae种内菌株的遗传分化很大程度上可能是与寄主间的互作进化有关,如侵染水稻的菌株在进化树分析中可聚为明显不同的一小类;王金生等研究也表明在水稻和玉米D. zeae菌株间未发现共同的抗原,且水稻菌株的致病力较玉米菌株强,寄主范围也较广[26]。本研究同样发现,来源广东的6个D. zeae菌株的遗传分化程度与它们的寄主间的亲缘关系类似;芭蕉芋菌株在进化树分析中与香蕉菌株MS1和MS2的亲缘关系较近,它们与另外的3个水稻菌株分别属于差异较大的两类菌株。因此,来源于同一地域(广东)的两类不同寄主(芭蕉芋和香蕉为一类,水稻为一类)的6个菌株,其比较基因组学分析对于研究与寄主互作相关的D. zeae种内病原菌的遗传分化机制具有较好的研究意义;同时,芭蕉芋细菌性软腐病菌D. zeae CE1与寄主亲缘关系、菌株遗传亲缘关系均较为相近的病原菌间的比较分析,也可为揭示该病害发生流行规律和风险预警提供基因组学理论依据。
以上6个广东菌株的基因家族分析表明,多数基因在6个菌株间均是保守的(图 3);细菌分泌系统、鞭毛、胞外多糖、CRISPR等重要致病因子相关基因并未被发现,仅是某个菌株所特有的。已有研究表明,T3SS、T4SS、T5SS是包括Dickeya病原菌在内的Pectobacteriaceae科细菌的重要致病因子[27-29]。然而,本研究发现T3SS、T4SS、T5SS在部分D. zeae菌株是缺失的,如水稻菌株EC2缺失3个分泌系统,芭蕉芋菌株CE1缺失T3SS和T4SS,香蕉菌株MS2则缺失了大部分的T4SS基因,因此这3个分泌系统并非是D. zeae菌株侵染寄主所必需的;同样地,T3SS、T4SS、T5SS在D. paradisiaca菌株中也是自然缺失的。相反地,T1SS、T2SS和T6SS在6个来源于广东的D. zeae菌株中均存在,它们在NCBI数据库上已公开的D. zeae种内其他菌株的基因组中也是保守的。T1SS分泌的金属蛋白酶主要作用于植物细胞壁蛋白或与修饰病原菌分泌酶的活性相关[30];T2SS则分泌可分解植物细胞壁的细胞壁降解酶[31],这些细胞壁降解酶是软腐病菌侵染寄主产生软腐症状的直接手段[32]。因此,T1SS、T2SS是该病原菌侵染寄主植物并产生软腐症状的重要致病因子,这可能也是T1SS、T2SS在D. zeae种内菌株保守存在的原因;再比较Dickeya属其他种的菌株,T1SS、T2SS在其他不同种中也是保守的,包括缺失了T3SS、T4SS、T5SS的D. paradisiaca种。T6SS则是革兰氏阴性菌中最新被鉴定的分泌系统[33],近年研究表明,该分泌系统在革兰氏阴性菌中是潜在的致病因子[34]。与Dickeya属细菌T6SS相关的研究,目前仅见到效应蛋白(Rhs)的研究,该研究表明Rhs蛋白的C-端毒性结构域具有核酸酶活性且可抑制相邻细胞生长[35],这与已有研究表明Ⅵ型分泌系统可介导细菌间拮抗机制[36]是一致的。因此,除了T1SS和T2SS可作用于植物细胞壁并产生软腐病症状外,T6SS在D. zeae菌株间保守可能是由于该细菌的致病进程需要其他不同于T1SS和T2SS的作用机制,以助于病原菌实现有效定殖、增殖及适应寄主内的新环境等[37]。
在上述这些细菌的重要致病因子中,细菌分泌系统在两类不同寄主来源的菌株中并未表现明显的遗传分化,胞外多糖基因簇和CRISPR序列同样也未表现明显的分化,有必要进一步探讨以上基因簇之外的基因组位点在不同类型菌株间的遗传分化。本研究发现,80个基因是芭蕉芋和香蕉菌株特有的,42个基因是水稻菌株所特有的,在这些特异基因中,部分基因是成簇分布的,这些基因可能行使相同或类似的基因功能。在芭蕉芋和香蕉菌株中发现了2个基因簇,第1个基因簇的3个基因都可比对上与病原菌-植物互作相关的功能基因,其中fabF编码β-酮脂酰ACP合成酶,该酶是细菌脂肪酸合成途径中的关键酶,被称为长链3-酮脂酰ACP合成酶[38]。脂肪酸合成代谢是细菌的重要生理功能,脂肪酸合成不仅与细胞膜磷脂的合成相关,其合成途径也与类脂A、群体感应信号分子等活性分子的合成相关[39],如fabF缺失可能会改变细胞的脂肪酸组成,进而影响铜绿假单胞菌的群集运动能力[40]。第2个基因簇基因的功能较为一致,它们可能与细菌的群体感应相关。群体感应系统可调控病原菌致病基因的表达及毒力因子的产生和分泌等,如在Dickeya属细菌中广泛存在的酰基高丝氨酸内酯(acylhomoserine lactone,AHL)群体感应系统主要由编码AHL合成酶的luxI和AHL信号受体的luxR基因构成,该信号系统与水稻细菌性软腐病菌的细胞运动性、细菌生物被膜形成以及致病力等相关[41]。在这个与群体感应信号系统相关的特异基因簇中,vanR是属于LuxR蛋白家族的转录调控因子,基因功能预测表明该基因可能与双组分系统和细菌生物被膜相关,并且可能与病原菌-植物互作相关。
与芭蕉芋、香蕉菌株特异基因的COG功能较多地预测为一般功能明显不同,水稻菌株特异基因的COG功能更多地预测为碳水化合物转运和代谢,该COG功能所包含的基因占所有水稻菌株特异基因的比例约为36% (15/42)。另外,19% (8/42)水稻菌株的特异基因可比对上碳水化合物活性酶数据库,明显高于可比对上该数据库的芭蕉芋和香蕉菌株的特异基因所占的比例,仅9% (7/80)的基因可比对上。以上结果表明,与碳水化合物转运和代谢相关的基因可能在水稻菌株和寄主互作间表现较为重要的作用,而且部分基因预测可能与碳水化合物活性酶相关,其中yulB-rbsA2-rbsC- lsrD-alsE-alsK-rbsK是这类基因中的一个典型基因簇。我们在type I-E CRISPR的邻接位点上发现了3个成簇分布的水稻菌株特异基因。CRISPR序列是由特殊的DNA重复序列和cas基因序列组成,它们可发挥保护细菌不受细噬菌体或外源质粒入侵的作用[42]。除了该免疫功能之外,CRISPR系统还参与了细菌其他许多重要的生理过程,如细菌毒力和细菌生物被膜形成等[43]。CRISPR也可能与调控细菌的基因组进化相关[44],有研究表明热袍菌目细菌的CRISPR序列存在于基因组重排片段的边界处[45]。因此,该位于Type I-E CRISPR边界处的水稻菌株特异基因簇的功能是否与病原菌-寄主互作相关值得进一步深入探讨。
使用9个保守基因串联序列作上述两类寄主来源菌株之间的基因流(gene flow)分析,结果表明两类菌株间的Fst=0.259 33,小于0.33,表明两类寄主来源的菌株间存在频繁的基因交流[46]。上述两类不同寄主来源的菌株间可能存在频繁的基因交流,而同一类寄主来源的菌株之间的核苷酸多态性更低,它们之间同理也应该存在频繁的基因交流。这些菌株可能来源于相同的祖先,在与植物互作过程中逐渐产生相应的遗传分化。本研究在两类不同寄主来源菌株的特异基因位点上,发现了一些预测与病原菌-植物互作相关或者位于特殊基因组区域的基因或者基因簇,它们可能是病原菌在与植物互作过程所获得的,但是它们的具体功能有待下一步研究。
可能由于抗性较强的原因,关于芭蕉芋病虫害发生的报道较少。在广东省内,D. zeae病原菌危害水稻、香蕉等其他单子叶植物的报道较多。植物细菌性软腐病菌D. zeae在与不同寄主互作时可能会产生遗传分化并进化出相应的适应机制;另一方面,芭蕉芋和香蕉等作物均是热带亚热带作物并隶属同一目,地理位置、植物学分类上均较为接近,这些可能是芭蕉芋受到D. zeae危害的重要原因。因此,本研究的结果可丰富芭蕉芋有害生物的基础性研究,引起生产上对该病害的重视,有利于芭蕉芋产业中对于该类细菌性病害的防控,减少病害损失;另外,与香蕉、水稻等重要作物亲缘关系较为接近的作物可能也存在受D. zeae病原菌侵染的风险,本研究中D. zeae病原菌与寄主互作相关的遗传分化研究可提供预警依据。
| [1] |
Adeolu M, Alnajar S, Naushad S, et al. Genome-based phylogeny and taxonomy of the'Enterobacteriales':proposal for Enterobacterales ord.nov.divided into the families Enterobacteriaceae, Erwiniaceae fam.nov., Pectobacteriaceae fam.nov., Yersiniaceae fam.nov., Hafniaceae fam.nov., Morganellaceae fam.nov., and Budviciaceae fam.nov.[J]. International Journal of Systematic and Evolutionary Microbiology, 2016, 66(12): 5575-5599. DOI:10.1099/ijsem.0.001485 |
| [2] |
Feng J. Recent advances in taxonomy of plant pathogenic bacteria[J]. Scientia Agricultura Sinica, 2017, 50(12): 2305-2314. (in Chinese) 冯洁. 植物病原细菌分类最新进展[J]. 中国农业科学, 2017, 50(12): 2305-2314. |
| [3] |
Lin BR, Shen HF, Pu XM, et al. First report of a soft rot of banana in mainland China caused by a Dickeya sp.(Pectobacterium chrysanthemi)[J]. Plant Disease, 2010, 94(5): 640. |
| [4] |
Zhang JX, Shen HF, Pu XM, et al. Identification of Dickeya zeae as a causal agent of bacterial soft rot in banana in China[J]. Plant Disease, 2014, 98(4): 436-442. DOI:10.1094/PDIS-07-13-0711-RE |
| [5] |
Yang QY, Jiang SB, Zhang JX, et al. First report of Dickeya zeae causing bacterial soft rot on Canna edulis in China[J]. Plant Disease, 2019, 103(1): 146. |
| [6] |
Zhou ZB. Development situation and potentiality of Canna edulis in Guizhou[J]. Guizhou Agricultural Sciences, 2009, 37(2): 136-137. (in Chinese) 周正邦. 贵州芭蕉芋发展现状及潜力分析[J]. 贵州农业科学, 2009, 37(2): 136-137. |
| [7] |
Liu Q, Fu P, Su M, et al. Preliminary report on collection and observation of Canna variety resources[J]. Seed, 2013, 32(12): 89-90, 101. (in Chinese) 柳茜, 傅平, 苏茂, 等. 芭蕉芋品种资源观察试验[J]. 种子, 2013, 32(12): 89-90, 101. |
| [8] |
Zhou MQ, Ou ZG, Liu FZ, et al. Comparative test of 12 forage Canna edulis varieties[J]. Chinese Journal of Tropical Crops, 2016, 37(9): 1677-1682. (in Chinese) 周明强, 欧珍贵, 刘凡值, 等. 12个饲用芭蕉芋品种(材料)比较试验[J]. 热带作物学报, 2016, 37(9): 1677-1682. |
| [9] |
Ou ZG, Zhou ZB, Zhou MQ. Research progress in germplasm and cultivation technique of Canna edulis[J]. Hubei Agricultural Sciences, 2012, 51(3): 441-445. (in Chinese) 欧珍贵, 周正邦, 周明强. 芭蕉芋的种质资源及栽培技术研究进展[J]. 湖北农业科学, 2012, 51(3): 441-445. |
| [10] |
Zhang AY, Xu ZL, Xu JC, et al. Study on the planting mode of corn interplanting with Canna edulis[J]. Serves of Agricultural Technology, 2008, 25(9): 16. (in Chinese) 张爱玉, 徐之龙, 徐俊昌, 等. 芭蕉芋间套作玉米种植模式研究[J]. 农技服务, 2008, 25(9): 16. |
| [11] |
Pritchard L, Humphris S, Saddler GS, et al. Draft genome sequences of 17 isolates of the plant pathogenic bacterium Dickeya[J]. Genome Announcements, 2013, 1(6): e00978-13. DOI:10.1128/genomeA.00978-13 |
| [12] |
Chin CS, Alexander DH, Marks P, et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data[J]. Nature Methods, 2013, 10(6): 563-569. DOI:10.1038/nmeth.2474 |
| [13] |
Myers EW, Sutton GG, Delcher AL, et al. A whole-genome assembly of Drosophila[J]. Science, 2000, 287(5461): 2196-2204. DOI:10.1126/science.287.5461.2196 |
| [14] |
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform[J]. Bioinformatics, 2009, 25(14): 1754-1760. DOI:10.1093/bioinformatics/btp324 |
| [15] |
Walker BJ, Abeel T, Shea T, et al. Pilon:an integrated tool for comprehensive microbial variant detection and genome assembly improvement[J]. PLoS One, 2014, 9(11): e112963. DOI:10.1371/journal.pone.0112963 |
| [16] |
Chen NS. Using RepeatMasker to identify repetitive elements in genomic sequences[J]. Current Protocols in Bioinformatics, 2004, 25: 4.10.1-4.10.14. |
| [17] |
Lagesen K, Hallin P, Rødland EA, et al. RNAmmer:consistent and rapid annotation of ribosomal RNA genes[J]. Nucleic Acids Research, 2007, 35(9): 3100-3108. DOI:10.1093/nar/gkm160 |
| [18] |
Lowe TM, Eddy SR. tRNAscan-SE:a program for improved detection of transfer RNA genes in genomic sequence[J]. Nucleic Acids Research, 1997, 25(5): 955-964. DOI:10.1093/nar/25.5.955 |
| [19] |
Besemer J, Lomsadze A, Borodovsky M. GeneMarkS:a self-training method for prediction of gene starts in microbial genomes.Implications for finding sequence motifs in regulatory regions[J]. Nucleic Acids Research, 2001, 29(12): 2607-2618. DOI:10.1093/nar/29.12.2607 |
| [20] |
Krzywinski M, Schein J, Birol I, et al. Circos:an information aesthetic for comparative genomics[J]. Genome Research, 2009, 19(9): 1639-1645. DOI:10.1101/gr.092759.109 |
| [21] |
Grissa I, Vergnaud G, Pourcel C. CRISPRFinder:a web tool to identify clustered regularly interspaced short palindromic repeats[J]. Nucleic Acids Research, 2007, 35(Web Server issue): W52-W57. |
| [22] |
Blin K, Wolf T, Chevrette MG, et al. antiSMASH 4.0-improvements in chemistry prediction and gene cluster boundary identification[J]. Nucleic Acids Research, 2017, 45(W1): W36-W41. DOI:10.1093/nar/gkx319 |
| [23] |
Li L, Stoeckert CJ Jr, Roos DS. OrthoMCL:identification of ortholog groups for eukaryotic genomes[J]. Genome Research, 2003, 13(9): 2178-2189. DOI:10.1101/gr.1224503 |
| [24] |
Parkinson N, Stead D, Bew J, et al. Dickeya species relatedness and clade structure determined by comparison of recA sequences[J]. International Journal of Systematic and Evolutionary Microbiology, 2009, 59(10): 2388-2393. DOI:10.1099/ijs.0.009258-0 |
| [25] |
Hu M, Li JL, Chen RT, et al. Dickeya zeae strains isolated from rice, banana and clivia rot plants show great virulence differentials[J]. BMC Microbiology, 2018, 18(1): 136. |
| [26] |
Wang JS, Yang XY. Comparative study on the causal bacteria of rice foot rod ant corn stalk rot[J]. Acta Phytopathologica Sinica, 1991, 21(3): 181-185. (in Chinese) 王金生, 杨晓云. 水稻基腐病细菌和玉米茎腐病细菌的比较研究[J]. 植物病理学报, 1991, 21(3): 181-185. |
| [27] |
Yang SH, Peng Q, San Francisco M, et al. Type Ⅲ secretion system genes of Dickeya dadantii 3937 are induced by plant phenolic acids[J]. PLoS One, 2008, 3(8): e2973. DOI:10.1371/journal.pone.0002973 |
| [28] |
Bell KS, Sebaihia M, Pritchard L, et al. Genome sequence of the enterobacterial phytopathogen Erwinia carotovora subsp.atroseptica and characterization of virulence factors[J]. Proceedings of the National Academy of Sciences of the United States of America, 2014, 101(30): 11105-11110. |
| [29] |
Rojas CM, Ham JH, Deng WL, et al. HecA, a member of a class of adhesins produced by diverse pathogenic bacteria, contributes to the attachment, aggregation, epidermal cell killing, and virulence phenotypes of Erwinia chrysanthemi EC16 on Nicotiana clevelandii seedlings[J]. Proceedings of the National Academy of Sciences of the United States of America, 2002, 99(20): 13142-13147. DOI:10.1073/pnas.202358699 |
| [30] |
Hommais F, Oger-Desfeux C, van Gijsegem F, et al. PecS is a global regulator of the symptomatic phase in the phytopathogenic bacterium Erwinia chrysanthemi 3937[J]. Journal of Bacteriology, 2008, 190(22): 7508-7522. DOI:10.1128/JB.00553-08 |
| [31] |
Kazemi-Pour N, Condemine G, Hugouvieux-Cotte-Pattat N. The secretome of the plant pathogenic bacterium Erwinia chrysanthemi[J]. Proteomics, 2004, 4(10): 3177-3186. DOI:10.1002/pmic.200300814 |
| [32] |
Toth IK, Birch PRJ. Rotting softly and stealthily[J]. Current Opinion in Plant Biology, 2005, 8(4): 424-429. DOI:10.1016/j.pbi.2005.04.001 |
| [33] |
Mougous JD, Cuff ME, Raunser S, et al. A virulence locus of Pseudomonas aeruginosa encodes a protein secretion apparatus[J]. Science, 2006, 312(5779): 1526-1530. DOI:10.1126/science.1128393 |
| [34] |
Hood RD, Singh P, Hsu F, et al. A type Ⅵ secretion system of Pseudomonas aeruginosa targets a toxin to bacteria[J]. Cell Host & Microbe, 2010, 7(1): 25-37. |
| [35] |
Koskiniemi S, Lamoureux JG, Nikolakakis KC, et al. Rhs proteins from diverse bacteria mediate intercellular competition[J]. Proceedings of the National Academy of Sciences of the United States of America, 2013, 110(17): 7032-7037. DOI:10.1073/pnas.1300627110 |
| [36] |
Russell AB, Peterson SB, Mougous JD. Type Ⅵ secretion system effectors:poisons with a purpose[J]. Nature Reviews Microbiology, 2014, 12(2): 137-148. DOI:10.1038/nrmicro3185 |
| [37] |
Reverchon S, Muskhelisvili G, Nasser W. Virulence program of a bacterial plant pathogen:the Dickeya model[J]. Progress in Molecular Biology and Translational Science, 2016, 142: 51-92. DOI:10.1016/bs.pmbts.2016.05.005 |
| [38] |
Campbell JW, Cronan JE Jr. Bacterial fatty acid biosynthesis:targets for antibacterial drug discovery[J]. Annual Review of Microbiology, 2001, 55(1): 305-332. |
| [39] |
Ma JR, Yu YH. Function of the long chain 3-ketoacyl-ACP synthase in Streptomyces coelicolor[J]. Microbiology China, 2019, 46(9): 2302-2311. (in Chinese) 马建荣, 余永红. 天蓝色链霉菌中长链3-酮脂酰ACP合成酶的功能[J]. 微生物学通报, 2019, 46(9): 2302-2311. |
| [40] |
Yu FF, Yu XY, Yu HY, et al. Effect of cis-vaccinate acid on swarming ability of Pseudomonas aeruginosa[J]. Acta Microbiologica Sinica, 2015, 55(12): 1600-1607. (in Chinese) 于非非, 余祥勇, 于海英, 等. 异油酸含量对铜绿假单胞菌群集运动能力的影响[J]. 微生物学报, 2015, 55(12): 1600-1607. |
| [41] |
Zhou JN, Jiang ZD, Zhang LH. Research progress in the pathogenic mechanism of Dickeya spp[J]. Acta Phytopathologica Sinica, 2015, 45(4): 337-349. (in Chinese) 周佳暖, 姜子德, 张炼辉. 细菌性软腐病菌Dickeya致病机理的研究进展[J]. 植物病理学报, 2015, 45(4): 337-349. |
| [42] |
Mojica FJM, Díez-villaseñor C, García-martínez J, et al. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements[J]. Journal of Molecular Evolution, 2005, 60(2): 174-182. DOI:10.1007/s00239-004-0046-3 |
| [43] |
Zhang B, Yao XP, Wang Y, et al. Advances in the mechanism of CRISPR-Cas system for regulating bacterial physiological function[J]. Microbiology China, 2017, 44(12): 3045-3052. (in Chinese) 张博, 姚学萍, 王印, 等. CRISPR-Cas系统调控细菌生理功能的机制研究进展[J]. 微生物学通报, 2017, 44(12): 3045-3052. |
| [44] |
Riehle MM, Bennett AF, Long AD. Genetic architecture of thermal adaptation in Escherichia coli[J]. Proceedings of the National Academy of Sciences of the United States of America, 2001, 98(2): 525-530. DOI:10.1073/pnas.98.2.525 |
| [45] |
Deboy RT, Mongodin EF, Emerson JB, et al. Chromosome evolution in the Thermotogales:large-scale inversions and strain diversification of CRISPR sequences[J]. Journal of Bacteriology, 2006, 188(7): 2364-2374. DOI:10.1128/JB.188.7.2364-2374.2006 |
| [46] |
Wei TY, Yang JG, Liao FL, et al. Genetic diversity and population structure of rice stripe virus in China[J]. Journal of General Virology, 2009, 90(4): 1025-1034. DOI:10.1099/vir.0.006858-0 |