 2020, Vol. 47
2020, Vol. 47扩展功能
文章信息
- 李海伟, 张法, 郑舰艇
- LI Hai-Wei, ZHANG Fa, ZHENG Jian-Ting
- 阿维菌素起始酰基转移酶的底物特异性
- Specificity of the loading acyltransferase from avermectin modular polyketide synthase Pharmaceutical Microbiology
- 微生物学通报, 2020, 47(1): 190-199
- Microbiology China, 2020, 47(1): 190-199
- DOI: 10.13344/j.microbiol.china.190236
-
文章历史
- 收稿日期: 2019-03-26
- 接受日期: 2019-05-21
- 网络首发日期: 2019-08-26

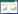




聚酮(polyketide)是一类由细菌、真菌和植物等产生的次级代谢产物,具有广泛的生物学和药理学活性,包括抗细菌、抗肿瘤、抗真菌、抗寄生虫和免疫抑制等。催化聚酮合成的酶称为聚酮合酶(polyketide synthase,PKS),根据催化中心的不同组织方式,PKS被划分成多种不同的类型,模块化PKS因结构域的模块化组织方式而得名[1-3]。聚酮化合物的生物合成类似于脂肪酸的生物合成,由酰基起始单元与一系列延伸单元缩合形成。与脂肪酸生物合成相比,模块化PKS能够利用更多种类起始和延伸单元,使其合成的聚酮化合物的结构更具多样性[4]。最小延伸模块具有3个结构域:酮合成酶(ketosynthase,KS)、酰基转移酶(acyltransferase,AT)和酰基载体蛋白(acyl carrier protein,ACP)。AT识别酰基底物并将其通过ACP转运至KS完成缩合反应。除此之外,一个模块中还可能含有酮还原酶(ketoreductase,KR)、烯酰基还原酶(enoylreductase,ER)和脱水酶(dehydratase,DH)。
阿维菌素(avermectins,Ave)及其衍生物是畜牧业、农业和人类传染病领域主要的抗寄生虫剂,由阿维链霉菌(Streptomyces avermitilis)发酵产生[5]。阿维菌素PKS是模块化的PKS,通过向起始单元上添加丙酸盐或乙酸盐形成阿维菌素起始糖苷配基(aglycone)[6]。起始糖苷配基经过一系列的修饰产生阿维菌素。在天然状况下,阿维菌素PKS的起始模块能够以2-甲基丁酰-CoA (2-methylbutyryl-CoA,MB-CoA)或异丁酰-CoA (isobutyryl-CoA,IB-CoA)作为起始单元分别合成“a”系列或“b”系列的阿维菌素(图 1),底物喂养实验发现阿维链霉菌突变株利用40种羧酸作为起始单元产生相应的衍生物,例如多拉菌素(doramectin)[7]。天然阿维菌素有8种组分,组分a所占比例较大,大约在80%以上,组分b则一般不超过20%,工业发酵生产过程不易将两者分离[8]。

|
| 图 1 阿维菌素PKS起始模块的AT Figure 1 Loading AT within avermectin PKS |
|
|
在许多大环内酯类抗生素的生物合成过程中,AT0充当“守门人”的作用,识别并且加载起始单元启动聚酮化合物的合成[9]。有些聚酮合酶的起始模块与阿维菌素的类似,只含有AT0和ACP0,有学者将其称为AVE型AT0[4]。与AVE型AT0不同,超过半数的PKS起始模块都含有一个类似于KS的N末端结构域,称为KSQ,此类AT0称为KSQ型AT0。KSQ与KS结构非常相似,只是催化活性位点的半胱氨酸被谷氨酰胺(Q)取代[10]。AVE型聚酮链的起始,是由AT0结合酰基-CoA底物,将酰基传递到下一模块的KS,一般有着较为宽泛的底物特异性,常见的有乙酰-CoA (acetyl-CoA,A-CoA)、丙酰-CoA (propionyl-CoA,P-CoA)、IB-CoA、MB-CoA和3-甲基丁酰-CoA (3-methylbutyryl-CoA,3-MB-CoA)等。KSQ型聚酮链的起始是通过KSQ催化AT0结合的二元羧酸的CoA进行脱羧反应,为聚酮化合物的合成提供乙酰或丙酸起始单元[11],有着严格的底物特异性,底物一般为丙二酰-CoA (malonyl-CoA,M-CoA)或者甲基丙二酰-CoA (methylmalonyl- CoA,MM-CoA)。与延伸模块的AT类似,KSQ型的AT0在其活性位点中含有保守的精氨酸[12]。常见的AVE型AT0有:识别MB-CoA而不识别IB-CoA的兰卡霉素(lankamycin,Lkm)的AT0[13];以IB-CoA为起始单元的泰托霉素(tautomycin,Tauto)和脂霉素(lipomycin,Lip)的AT0[14-15]。常见的KSQ型AT0有:以M-CoA为起始单元的多杀菌素(spinosyn)[16]的AT0;以MM-CoA为起始单元的莫能菌素(monensin)的AT0[17]。
实验使用结构较简单的酰基-N-乙酰半胱胺(N-acetylcysteamine,SNAC)即MB-SNAC和IB-SNAC为底物替代价格较昂贵的MB-CoA和IB-CoA进行实验[18]。本研究中将识别不同底物的AT0进行氨基酸序列比对,找到可能影响Ave AT0底物选择性的关键氨基酸,并对其进行定点突变,得到单突变体AveAT0 V224M、双突变体AveAT0 V224M/Q149L和三突变体AveAT0 V224M/Q149L/L121M。通过测量突变体的水解活性和酶动力学常数,发现三突变对两种底物的偏好性增大。本研究不仅得到了底物偏好性改变的突变体,还为定向改造阿维菌素PKS的产物提供了依据。
1 材料与方法 1.1 材料 1.1.1 菌株、质粒及引物Escherichia coli BL21(DE3)、E. coli DH10B及重组质粒pET-28a-AveAT0均为本实验室保藏。引物如表 1所示,由上海捷瑞生物工程有限公司合成。
| 引物 Primers |
序列 Sequences (5′→3′) |
| V224M-F | CGCGCATGATCCCGGTGGACATGCCCGCCCACTCCCCCCTG |
| V224M-R | GTCCACCGGGATCATGCGCGTGCGCACCTGCGCGGCGG |
| Q149L-F | TGACGCTTTGGAGCCAGGCACTGACCACCCTTGCCGGGACC |
| Q149L-R | TGCCTGGCTCCAAAGCGTCACCACGCGTGCGGCGTCGG |
| L121M-F | GCGCGGTGCTGGGACACAGCATGGGCGAGATCGCGGCAGCC |
| L121M-R | GCTGTGTCCCAGCACCGCGCACGGCTCGACCCCTTGCG |
限制性内切酶,Thermo Fisher Scientific公司;质粒提取试剂盒、PCR产物纯化试剂盒、胶回收试剂盒,生工生物工程(上海)股份有限公司;试剂N-乙酰半胱氨酸(N-acetylcysteamine,SNAC)、5, 5′-二硫代双(2-硝基苯甲酸) [5, 5′-dithiobis-(2-nitrobenzoic acid),DTNB],上海阿拉丁生化科技股份有限公司;2-甲基丁酰-SNAC和异丁酰-SNAC合成及表征已在实验室前期论文[4]中发表。
1.1.3 主要仪器超微量核酸蛋白分析仪NanoDrop 2000,Thermo Fisher Scientific公司;快速蛋白液相色谱(fast protein liquid chromatography,FPLC)、聚合酶链式核酸扩增仪,Bio-Rad公司;高速冷冻离心机,Eppendorf公司;生化培养箱,上海一恒科学仪器有限公司。
1.2 方法 1.2.1 AT0序列分析从NCBI查询得到各种AT0序列,使用软件ClustalX进行序列相似性分析,并运用MEGA 5.05邻接法构建进化树。
1.2.2 定点突变定点突变初始以pET-28a-AveAT0为模板,以表 1中的V224M-F和V224M-R为引物进行PCR扩增。PCR反应体系(50 μL):2×Phanta Max Buffer 25 μL,ddH2O 19.6 μL,二甲基亚砜(dimethyl sulfoxide,DMSO) 2.0 μL,dNTPs (10 mmol/L) 1 μL,模板(100 ng/μL) 1 μL,上、下游引物(100 µmol/L)各0.2 μL,Phanta Max Super-Fidelity DNA Polymerase (1 U/μL) 1 μL。PCR反应条件:95 ℃ 3 min;95 ℃ 15 s,60 ℃ 15 s,72 ℃ 6 min,25个循环;72 ℃ 10 min。PCR产物经Dpn I内切酶37 ℃消化处理1.5 h后,转化进E. coli DH10B感受态细胞,测序正确后得到单突变质粒pET-28a-AveAT0 V224M。以pET-28a-AveAT0 V224M为模板,表 1中的Q149L-F、Q149L-R为引物构建双突变质粒pET-28a-AveAT0 V224M/Q149。最后,以类似的方法构建三突变质粒pET-28a- AveAT0 V224M/Q149L/L121M。
1.2.3 蛋白表达纯化将构建好的表达载体质粒转化进E. coli BL21(DE3)感受态细胞,37 ℃培养过夜。挑取转化子于50 mL LB (卡那霉素,50 μg/mL)培养基,37 ℃、220 r/min培养6−8 h。按1%的接种量转接到1 L (卡那霉素,50 μg/mL) LB培养基中,37 ℃、220 r/min培养约2−3 h至OD600为0.4时,将摇床降温至16 ℃,继续培养至OD600为0.6时加入IPTG至终浓度为0.15 mmol/L,16 ℃培养14−16 h。4 ℃、3 000×g离心10 min收集菌体,结合缓冲溶液[50 mmol/L Tris,500 mmol/L NaCl,10% (质量体积比)甘油,pH 7.5]重悬,超声破碎菌体。4 ℃、13 000×g离心45 min除去细胞碎片。上清液倒入镍亲和层析柱中,冲洗缓冲液(结合缓冲液加15 mmol/L咪唑)除去杂蛋白,洗脱缓冲液(结合缓冲液加300 mmol/L咪唑)将目的蛋白洗出。将初步纯化的蛋白用分子排阻FPLC (SuperdexTM 200 Increase 10/300 GL层析柱)进行进一步纯化,除去多聚体和咪唑。纯化后的蛋白用SDS-PAGE检测,确定分子量及纯度。
1.2.4 生化反应AT能够将酰基-SNAC和酰基-CoA水解,得到游离的SNAC和CoA。CoA和SNAC含有巯基,可以用Ellman测试法检测。含巯基化合物与DTNB反应,DTNB的二硫键断裂,产生2-硝基- 5-硫代苯甲酸(TNB−),在中性或碱性pH条件下离子化,生成TNB2−二价阴离子。这种TNB2−离子呈现黄色,在412 nm下有吸收峰。反应液加等体积的DMSO终止反应,之后加入等体积的DTNB溶液(2 mmol/L,0.1 mol/L Tris,pH 7.5)室温反应10 min,然后用NanoDrop在412 nm下测量的吸光度值。
反应速度v=(OD412/(14 150× 1)× 4× 106)/t
式中,OD412为减去对照的吸光值,t为反应时间[19]。
1.2.5 AveAT0及突变体水解活性的测定反应体系如下:5 μmol/L AveAT0,2 mmol/L的SNAC底物,10%甘油,10 mmol/L Tris,150 mmol/L NaCl,pH 7.5。37 ℃反应5 min后加入等体积的DMSO终止反应,之后用Ellman测试法检测OD412并求出AveAT0的反应速度v和v/E0 (E0为AT的浓度)。每个反应设置3个平行,将上述体系中的酶及底物换成相应缓冲液,其余保持相同作为对照。按照上述方法进一步测定单突变AveAT0 V224M、双突变AveAT0 V224M/Q149L和三突变AveAT0 V224M/Q149L/L121M的v/E0。
1.2.6 动力学参数的测定改变待测底物的浓度,其他反应条件与1.2.5相同,底物浓度从15 μmol/L增加至2 000 μmol/L。将反应初速度利用软件GraphPad Prism 6拟合到米氏公式,得出相应底物的动力学参数。
2 结果与分析 2.1 AT0序列比对与定点突变AveAT0与底物MB-SNAC建模得到了识别底物起关键作用的13个氨基酸(Gln35,Gln92,His119,Ser120,Leu121,Trp145,Gln149,Leu158,Ile220,Val222,Val224,Ala226和His227)[4]。使用软件ClustalX针对AT0的13个残基进同源性序列比对(图 2)。大多数KSQ型AT0以M-CoA或MM-CoA为起始单元。与KSQ型AT0相比,AVE型AT0的底物识别残基相对不保守。KSQ型AT0在底物结合口袋的末端含有精氨酸(145位点,以AveAT0编号)和保守的“HAFH”或“YASH”模序(224−227位点)。精氨酸起到稳定羧酸基团的作用,“HAFH”或“YASH”帮助丙二酰基或者甲基丙二酰基结合。精氨酸侧链与二羧酸硫酯的离子相互作用对底物的结合施加了强烈的空间限制,使得KSQ型AT0有着严格的底物特异性。针对AveAT0在底物识别过程中起重要作用的13个氨基酸,将AveAT0序列与只识别MB-CoA的LkmAT0、只识别IB-CoA的TautoAT0和LipAT0的序列进行比对,发现Val224、Gln149、Leu121可能是影响AveAT0底物选择关键的氨基酸。

|
| 图 2 AVE型AT0与KSQ型AT0的序列比对 Figure 2 Amino acid sequence alignments of AVE-type AT0 and KSQ-type AT0 注:序列比对是根据AveAT0的编号.在底物识别时与酰基直接作用的氨基酸残基用“*”标记;保守的HFAH/YASH和精氨酸以及突变位点用黑线标出. Note: The alignment was numbered according to AveAT0. The residues which directly contacted the acyl moiety of the substrate were labeled by "*"; The conserved HFAH/YASH motifs and arginine residue, as well as the mutation sites were underlined by black line. |
|
|
系统发育树显示识别M-CoA的KSQ型AT0属于同一分支,有着较高的亲缘性;部分AVE型AT0和识别MM-CoA的KSQ型AT0有着一定的亲缘性,少部分AVE型AT0独自属于一个分支,与其他AT0亲缘关系低(图 3)。

|
| 图 3 AVE型AT0与KSQ型AT0的系统发育树 Figure 3 The phylogenetic tree of AVE-type AT0 and KSQ-type AT0 注:发育树依次列出了蛋白的名称、NCBI的登录号、生物合成的起始单元;标尺刻度:10%的序列差异;酰基-CoA:底物特异性为此酰基-CoA的AT0. Note: The phylogenetic tree listed in turn protein names, NCBI accession numbers and starter units for each of the AT0 domains; Bar: 10% sequence divergence; Acyl-CoA: Acyl-CoA specific AT0. |
|
|
将成功构建的重组质粒pET-28a-AveAT0或其突变体导入大肠杆菌E. coli BL21(DE3)中,诱导表达并收集菌体,通过镍柱亲和层析纯化,并利用快速蛋白液相色谱(FPLC)进一步去除多聚体和咪唑,最终得到的蛋白进行SDS-PAGE验证(图 4A),发现在约40 kD处有一条明显的蛋白条带,与理论大小一致[4],FPLC也在约40 kD处有单一的吸收峰(图 4B),确定为目的蛋白,表达量约为8 mg/L。

|
| 图 4 AveAT0 SDS-PAGE电泳(A)和FPLC分析(B) Figure 4 SDS-PAGE analysis (A) and FPLC of AveAT0 (B) |
|
|
为了检测定点突变对AveAT0水解活性的影响,以MB-SNAC和IB-SNAC为底物,在酶浓度已知的情况下,通过特定的底物浓度(2 mmol/L)测得AveAT0及各个突变体对MB-SNAC和IB-SNAC的v/E0。从图 5中可以看出,各个突变体对MB-SNAC和IB-SNAC的水解活性均有较大程度的下降,三突变AveAT0 V224M/Q149L/L121M活性下降得最少。AveAT0对MB-SNAC的v/E0值是IB-SNAC的2.6倍,而三突变AveAT0 V224M/Q149L/L121M对MB-SNAC的v/E0值是IB-SNAC的8.7倍(图 5)。说明三突变AveAT0 V224M/Q149L/L121M在底物浓度为2 mmol/L时对MB-SNAC的偏好性比AveAT0强。

|
| 图 5 AveAT0及突变体的水解活性 Figure 5 Rates of hydrolysis by AveAT0 and mutants |
|
|
为了进一步比较AveAT0和三突变AveAT0 V224M/Q149L/L121M对两种底物MB-SNAC和IB-SNAC的偏好程度,分别测定了以MB-SNAC和IB-SNAC为底物时两个酶的动力学参数(表 2)。AveAT0对MB-SNAC的kcat/Km值为32.1 L/(mmol·min),是对IB-SNAC [kcat/Km为7.5 L/(mmol·min)]的4.3倍;而三突变AveAT0 V224M/Q149L/L121M对MB-SNAC的kcat/Km值为6.9 L/(mmol·min),是对IB-SNAC为底物[kcat/Km为0.1 L/(mmol·min)]的60多倍。结果显示,三突变AveAT0 V224M/Q149L/L121M对MB-SNAC和IB-SNAC的kcat/Km差异是突变前的10倍以上,对MB-SNAC的偏好性较突变前有提高。
| AT domain | Substrate | kcat/Km (L/(mol·min)) | kcat (min−1) | Km (mmol/L) |
| AveAT0 | Isobutyryl SNAC | 7.5±1.9 | 6.4±0.6 | 0.8±0.2 |
| 2-Methylbutyryl SNAC | 32.1±3.9 | 14.1±0.6 | 0.4±0.05 | |
| V224M/Q149L/L121M | Isobutyryl SNAC | 0.1±0.03 | ND | ND |
| 2-Methylbutyryl SNAC | 6.9±2.1 | 5.4±0.7 | 0.8±0.2 | |
| 注:ND:未测量出. Note: ND: Not determined. |
||||
聚酮是已知的结构最多样化的分子之一,因具有抗菌、免疫抑制和抗癌等生物活性,其合成机制一直是研究的热点。模块化的PKS是研究最多和研究最透彻的PKS,对其进行工程化的改造有可能产生各种新颖的化合物。运用较多的方法是进行整个模块的交换,将其他PKS的模块交换于特定的PKS起始模块以及延伸模块中[20]。阿维菌素起始模块成功地被引入到红霉素聚酮合酶(deoxyerythronolide B synthase,DEBS)的第一个模块中,得到杂交的PKS基因在Saccharopolyspora erythraea中成功表达[21]。也有研究者用来自磷霉素的独特的环己烷羧酸(cyclohexanecarboxylic,CHC)起始模块替换了阿维链霉菌中阿维菌素PKS的起始模块,成功地改造出了多拉菌素的替代品[22-23]。因为AT的底物特异性决定了聚酮化合物的侧链结构,使得对AT结构域的改造成为近些年结构生物学研究的热点,常用的方法是交换识别不同底物的AT和定点突变。例如,用NidAT5 (niddamycin PKS第5模块的AT,特异性识别乙基丙二酰-CoA)替换红色糖多孢菌中DEBS上的EryAT4 (erythromycin PKS第4模块的AT,特异性识别MM-CoA),则合成了C-6位乙基化的红霉索类似物[24]。与交换AT结构域的改造方法相比,定点突变因是最小程度地对PKS进行改变,最大限度地减少了对蛋白-蛋白相互作用的有害干扰,因而成为更合理工程化改造AT的方法。
本实验结合了结构生物学、生物信息学和分子生物学的方法进行研究,以达到对AveAT0的底物特异性进行定向的改造。根据已解出的AveAT0的结构及与底物识别起关键作用的13个氨基酸,我们将识别不同底物的AT0进行氨基酸序列对比,找到了在底物的识别上关键的氨基酸,并据此对AveAT0进行了定点突变。最终得到了单突变体AveAT0 V224M、双突变体AveAT0 V224M/Q149L和三突变体AveAT0 V224M/Q149L/ L121M。单突变体和双突变体对MB-SNAC和IB-SNAC的水解活性均远低于AveAT0的10%,三突变对IB-SNAC的水解活性远低于AveAT0的10%,但对MB-SNAC的水解活性可达到AveAT0的30%。三突变对IB-SNAC的kcat/Km值是AveAT0的1.3%,但对MB-SNAC的kcat/Km值可达到AveAT0的21.5%,对MB-SNAC的偏好性较AveAT0有所提高。
目前工业生产的阿维菌素有8种组分。AveAT0识别MB-SNAC生成“a”系列,识别IB-SNAC生成“b”系列,工业发酵生产过程不易将二者分离[8]。本实验的研究结果对改造阿维菌素PKS,使其定向识别预期启动单元MB-CoA,生成特定的“a”组分的阿维菌素提供了理论基础。之后的工作中,我们计划将得到的对底物识别起重要作用的位点引入到阿维链霉菌体内,以求获得产生“a”组分比例更高的阿维菌素突变菌株。本研究对于提高阿维菌素“a”组分的生产比例,降低纯化成本、商业化生产阿维菌素提供了潜在的价值。
| [1] |
Khosla C, Herschlag D, Cane DE, et al. Assembly line polyketide synthases:mechanistic insights and unsolved problems[J]. Biochemistry, 2014, 53(18): 2875-2883. DOI:10.1021/bi500290t |
| [2] |
Xu W, Qiao KJ, Tang Y. Structural analysis of protein-protein interactions in type I polyketide synthases[J]. Critical Reviews in Biochemistry and Molecular Biology, 2013, 48(2): 98-122. DOI:10.3109/10409238.2012.745476 |
| [3] |
Dutta S, Whicher JR, Hansen DA, et al. Structure of a modular polyketide synthase[J]. Nature, 2014, 510(7506): 512-517. DOI:10.1038/nature13423 |
| [4] |
Wang F, Wang YJ, Ji JJ, et al. Structural and functional analysis of the loading acyltransferase from avermectin modular polyketide synthase[J]. ACS Chemical Biology, 2015, 10(4): 1017-1025. DOI:10.1021/cb500873k |
| [5] |
Yoon YJ, Kim ES, Hwang YS, et al. Avermectin:biochemical and molecular basis of its biosynthesis and regulation[J]. Applied Microbiology and Biotechnology, 2004, 63(6): 626-634. DOI:10.1007/s00253-003-1491-4 |
| [6] |
Zhuo Y, Zhang T, Wang Q, et al. Synthetic biology of avermectin for production improvement and structure diversification[J]. Biotechnology Journal, 2014, 9(3): 316-325. DOI:10.1002/biot.201200383 |
| [7] |
Dutton CJ, Gibson SP, Goudie AC, et al. Novel avermectins produced by mutational biosynthesis[J]. The Journal of Antibiotics, 1991, 44(3): 357-365. DOI:10.7164/antibiotics.44.357 |
| [8] |
Shoop WL, Mrozik H, Fisher MH. Structure and activity of avermectins and milbemycins in animal health[J]. Veterinary Parasitology, 1995, 59(2): 139-156. |
| [9] |
Crawford JM, Vagstad AL, Whitworth KP, et al. Synthetic strategy of nonreducing iterative polyketide synthases and the origin of the classical "starter-unit effect"[J]. ChemBioChem, 2008, 9(7): 1019-1023. DOI:10.1002/cbic.200700702 |
| [10] |
Bisang C, Long PF, Corte's J, et al. A chain initiation factor common to both modular and aromatic polyketide synthases[J]. Nature, 1999, 401(6752): 502-505. DOI:10.1038/46829 |
| [11] |
Long PF, Wilkinson CJ, Bisang CP, et al. Engineering specificity of starter unit selection by the erythromycin-producing polyketide synthase[J]. Molecular Microbiology, 2002, 43(5): 1215-1225. DOI:10.1046/j.1365-2958.2002.02815.x |
| [12] |
Oefner C, Schulz H, D'Arcy A, et al. Mapping the active site of Escherichia coli malonyl-CoA-acyl carrier protein transacylase (FabD) by protein crystallography[J]. Acta Crystallographica Section D:Biological Crystallography, 2006, 62(6): 613-618. DOI:10.1107/S0907444906009474 |
| [13] |
Arakawa K, Kodama K, Tatsuno S, et al. Analysis of the loading and hydroxylation steps in lankamycin biosynthesis in Streptomyces rochei[J]. Antimicrobial Agents and Chemotherapy, 2006, 50(6): 1946-1952. DOI:10.1128/AAC.00016-06 |
| [14] |
Li WL, Ju JH, Rajski SR, et al. Characterization of the tautomycin biosynthetic gene cluster from Streptomyces spiroverticillatus unveiling new insights into dialkylmaleic anhydride and polyketide biosynthesis[J]. The Journal of Biological Chemistry, 2008, 283(42): 28607-28617. DOI:10.1074/jbc.M804279200 |
| [15] |
Bihlmaier C, Welle E, Hofmann C, et al. Biosynthetic gene cluster for the polyenoyltetramic acid α-lipomycin[J]. Antimicrobial Agents and Chemotherapy, 2006, 50(6): 2113-2121. DOI:10.1128/AAC.00007-06 |
| [16] |
Waldron C, Matsushima P, Rosteck Jr PR, et al. Cloning and analysis of the spinosad biosynthetic gene cluster of Saccharopolyspora spinosa[J]. Chemistry & Biology, 2001, 8(5): 487-499. |
| [17] |
Leadlay PF, Staunton J, Oliynyk M, et al. Engineering of complex polyketide biosynthesis-insights from sequencing of the monensin biosynthetic gene cluster[J]. Journal of Industrial Microbiology and Biotechnology, 2001, 27(6): 360-367. DOI:10.1038/sj.jim.7000204 |
| [18] |
Dong SS, Wang YJ, Ji JJ, et al. Heterologous expression and characterization of a thermostable acyl-CoA synthetase[J]. Acta Microbiologica Sinica, 2016, 56(9): 1477-1485. (in Chinese) 董爽爽, 王衍杰, 季俊杰, 等. 热稳定酯酰辅酶A合成酶的异源表达及酶学特性[J]. 微生物学报, 2016, 56(9): 1477-1485. |
| [19] |
Riener CK, Kada G, Gruber HJ. Quick measurement of protein sulfhydryls with Ellman's reagent and with 4, 4′-dithiodipyridine[J]. Analytical and Bioanalytical Chemistry, 2002, 373(4/5): 266-276. |
| [20] |
Musiol-Kroll EM, Wohlleben W. Acyltransferases as tools for polyketide synthase engineering[J]. Antibiotics, 2018, 7(3): 62. DOI:10.3390/antibiotics7030062 |
| [21] |
Marsden AFA, Wilkinson B, Cortés J, et al. Engineering broader specificity into an antibiotic-producing polyketide synthase[J]. Science, 1998, 279(5348): 199-202. DOI:10.1126/science.279.5348.199 |
| [22] |
Palaniappan N, Kim BS, Sekiyama Y, et al. Enhancement and selective production of phoslactomycin B, a protein phosphatase IIa inhibitor, through identification and engineering of the corresponding biosynthetic gene cluster[J]. The Journal of Biological Chemistry, 2003, 278(37): 35552-35557. DOI:10.1074/jbc.M305082200 |
| [23] |
Ghatge M, Palaniappan N, Das Choudhuri S, et al. Genetic manipulation of the biosynthetic process leading to phoslactomycins, potent protein phosphatase 2A inhibitors[J]. Journal of Industrial Microbiology and Biotechnology, 2006, 33(7): 589-599. DOI:10.1007/s10295-006-0116-1 |
| [24] |
Stassi DL, Kakavas SJ, Reynolds KA, et al. Ethyl-substituted erythromycin derivatives produced by directed metabolic engineering[J]. Proceedings of the National Academy of Sciences of the United States of America, 1998, 95(13): 7305-7309. DOI:10.1073/pnas.95.13.7305 |