 2019, Vol. 46
2019, Vol. 46扩展功能
文章信息
- 余泽晖, 梁志伟, 李浩聪, 鲁祺鸿, 汪善全
- YU Ze-Hui, LIANG Zhi-Wei, LI Hao-Cong, LU Qi-hong, WANG Shan-Quan
- 厌氧消化污泥微生物组研究方法及应用
- Anaerobic sludge digestion microbiome ― analytical methods and applications
- 微生物学通报, 2019, 46(8): 2053-2068
- Microbiology China, 2019, 46(8): 2053-2068
- DOI: 10.13344/j.microbiol.china.190332
-
文章历史
- 收稿日期: 2019-04-15
- 接受日期: 2019-05-23
- 网络首发日期: 2019-06-05





2. 中山大学环境微生物组学研究中心 广东 广州 510006;
3. 广东省环境污染控制与修复技术重点实验室 广东 广州 510006
2. Environmental Microbiome Research Center, Sun Yat-Sen University, Guangzhou, Guangdong 510006, China;
3. Guangdong Provincial Key Laboratory of Environmental Pollution Control and Remediation Technology, Guangzhou, Guangdong 510006, China
市政污水处理过程中产生大量的剩余污泥,具有含水率高、有恶臭、难处理等特点,并且剩余污泥存在病原体、重金属等有害物质,如果不进行稳定化处理和安全处置会对环境造成危害[1]。目前如何实现高效的污泥减量化、稳定化处理处置问题已经成为环境保护及人居安全保障的一大难题。中国的待处理污泥总量在2010年就已经达到3 000万t (80%含水率),且每年以高于13%的增长率递增,但污泥安全处置率却低于35%[2]。在污泥的处理处置方法中,直接陆地填埋和焚烧等处理处置方法很难平衡土地空间需求、运营成本及环境保护法规间的需求[3]。污泥厌氧消化处理是实现剩余污泥减量化、稳定化的主要生物处理过程,并且可以通过产生甲烷回收部分能源。因此,污泥厌氧消化成为市政污水处理厂污泥减量及稳定化处理的核心环节。污泥消化反应器中复杂微生物组是维持污泥厌氧消化稳定运行和高效产气的关键[4-6]。因此,了解并调控厌氧消化污泥微生物组是保障污泥厌氧消化效率的前提和基础。
厌氧消化污泥微生物组是污泥厌氧消化反应器中所有微生物种群及其基因组的集合。通过深入了解厌氧消化污泥中微生物的多样性、结构、功能、互作关系,以及它们与反应器运行条件和微生物生态系统功能之间的关系,可以把握并调控剩余污泥中复杂有机物水解、发酵、产酸、产乙酸及产甲烷关键步骤,实现消化污泥种群结构与有机物产甲烷效率的优化。近20年来,高通量测序与生物信息处理技术的快速发展极大地促进了厌氧消化污泥微生物组的研究。尤其在生物信息学领域,通过结合数据挖掘、统计及机器学习等人工智能分析技术,极大促进了我们对污泥厌氧消化微生物组的认识。但由于厌氧消化污泥本身具有复杂的微生物种群组成(多样性及丰度)、错综复杂的种群共生关系(互作关系)及含有大量不可培养微生物(关键种群生理生化特征)等特点,解析消化污泥微生物组成为污泥厌氧消化研究及应用的瓶颈。
本文将主要从以下4个方面探讨厌氧消化污泥微生物组的研究及应用:(1)剩余活性污泥结构、组成及其厌氧消化;(2)基于16S rRNA基因序列测序的微生物组研究;(3)基于宏基因组及宏转录组分析的微生物组研究;(4)厌氧消化污泥微生物组研究案例分析。最后,我们提出了厌氧消化污泥微生物组研究需要解决的关键科学问题,并进一步展望了厌氧消化污泥微生物组未来的研究及应用。
1 剩余活性污泥结构、组成及厌氧消化 1.1 剩余活性污泥结构与组成市政污水处理厂产生的剩余污泥主要由初沉污泥及二沉剩余活性污泥组成。其中剩余活性污泥较初沉污泥可厌氧消化性低,并主要由微生物细胞、胞外多聚物(Extracellular polymeric substances,EPS)、可溶性微生物产物(Soluble microbial products,SMP)及无机颗粒等复杂成分组成[7-9](图 1A)。因此,剩余活性污泥结构、组成及其厌氧消化是本文讨论的重点。据统计,剩余活性污泥中微生物细胞大约占剩余活性污泥总量的10%-25%[10],主要包括病毒、细菌、真菌及原后生动物等,其中活体细胞占细胞总数的80%左右[11]。由脂类、多糖及蛋白质等形成的EPS是污泥组成中占比最高的组分,可进一步细分为紧密结合型EPS (TB-EPS)、松散结合型EPS (LB-EPS)和溶解型EPS (S-EPS)。TB-EPS附着在剩余活性污泥微生物细胞表面,并与细胞间存在物质交换;LB-EPS是由TB-EPS延伸而来的分散黏液层,具有高孔隙度和分散性结构,没有明显边缘;而S-EPS在絮体污泥间隙水相中均匀分布。EPS中有机大分子与微生物细胞死亡释放出的大分子及污水中其它复杂组分可进一步形成结构更为复杂、稳定的腐殖质并结成网状结构。污水有机污染物降解过程中,絮状活性污泥EPS为其微生物细胞提供了缓冲保护、应急碳源或能源及改善污泥絮凝沉淀效果等有益功能[12-14],但也因为EPS稳定、复杂的絮状结构制约了活性污泥中有机物的释放,成为污泥厌氧消化过程限制消化效率的瓶颈问题。

|
| 图 1 活性污泥絮体结构示意图(A)及国内各地区活性污泥有机物比例(B) Figure 1 Schematic diagram of activated sludge floc structure (A) and proportion of organic matter in activated sludge from different regions of China (B) 注:S-EPS:溶解型胞外聚合物;LB-EPS:松散结合型胞外聚合物;TB-EPS:紧密结合型胞外聚合物. Note: S-EPS: Slime EPS; LB-EPS: Loosely bound EPS; TB-EPS: Tightly bound EPS. |
|
|
微生物细胞及EPS等是活性污泥中有机物的主要组成。这些有机物(Volatile solids,VS)在总污泥固体(Total solids,TS)中占比(VS/TS)可以达到60%-80% (图 1B),并且与活性污泥的可厌氧消化性成正相关关系。在组成元素上,活性污泥主要由C、H、O、N元素组成,其比例与活性污泥经验分子式(C5H7O2N)基本吻合[15]。此外,剩余活性污泥絮体还含有大量P元素及丰富的金属元素,包括Na、Mg、Al、K、Ca、Fe等。
1.2 剩余活性污泥厌氧消化传统厌氧消化过程包括水解、发酵酸化、乙酸化及甲烷化4个阶段(图 2)[16]。在水解阶段,多糖、蛋白及脂类等大分子有机物在水解酶作用下转化为糖类、氨基酸及脂肪酸等小分子有机物。其中水解酶是发酵菌等功能种群分泌的胞外酶,因此水解过程是胞外生物转化过程。在酸化阶段,水解形成的简单结构有机物进入发酵菌细胞内部进一步转化为更小分子的有机酸及醇类。由于该过程在热动力学及反应动力学上都很容易进行,因此发酵产酸过程涉及的种群及生物量也最多最高。在产乙酸阶段,发酵产生的挥发性有机酸可以进一步由产乙酸菌转化为乙酸。从热动力学角度考虑,产乙酸过程可提供微生物代谢的能量有限,需要后续产甲烷菌不断消耗乙酸来配合该反应过程的顺利进行。最后,在产甲烷阶段,发酵及产乙酸过程形成的氢气、二氧化碳及乙酸可以由嗜氢型及嗜乙酸型产甲烷菌转化为甲烷。因此,整个厌氧消化反应需要以下4个大的功能种群形成紧密、互利共赢的协作关系来共同完成:(1)水解发酵产酸菌,包括梭菌(Clostridium)、拟杆菌(Bacteroides)、发酵乳杆菌(Lactobacillus)及地杆菌(Geobacter)等[17-18];(2)产乙酸菌,包括互营杆菌(Syntrophobacter)、互营单胞菌(Syntrophomonas)及互营热菌(Syntrophothermus)[19];(3)嗜氢型产甲烷菌,包括甲烷杆菌(Methanobacterium)及甲烷囊菌(Methanoculleus)等;(4)嗜乙酸型产甲烷菌,包括甲烷八叠球菌属(Methanosarcina)和甲烷丝菌属(Methanosaeta)等。

|
| 图 2 污泥厌氧消化主要步骤 Figure 2 Main steps of anaerobic sludge digestion |
|
|
剩余活性污泥中的有机固体(VS)非常顽固[20-21]。例如,活性污泥VS中,不可生物降解的腐殖质含量高达20%左右[10]。考虑到其它难降解有机物的存在,活性污泥中可厌氧消化的有机物不到活性污泥总有机物(VS)的70%。与传统厌氧消化过程相比,污泥厌氧消化在水解前需要解决的主要问题在于,如何高效破坏EPS及其包裹的微生物细胞结构并释放有机大分子(图 2)。因此,活性污泥中大分子有机物释放过程是污泥厌氧消化过程的速度和效率的限制步骤。实际污泥厌氧消化工程中通常会采用加热、加酸碱药剂等物理化学方法来促进有机物的释放过程,提高污泥可消化性[22-23]。在微生物群落结构与功能层面,厌氧消化污泥微生物组主要种群组成与传统厌氧消化反应器微生物组结构及功能相似,但由于污泥消化反应器进料主要是细胞分泌及本身的生物大分子,进料底物的不同导致微生物群落结构与功能有所不同。而且,由于受到活性污泥进料中兼氧及厌氧菌的干扰,污泥厌氧消化反应器中微生物组分析仍需清晰界定消化污泥微生物组中核心功能种群、冗余种群及其它种群。
2 厌氧消化污泥微生物组研究方法厌氧消化污泥微生物组由于其结构及功能复杂,对分析技术及方法要求较高。通常厌氧消化污泥微生物组的分子生物学分析包括以下2个大的部分(图 3):(1)基于DNA测序的微生物组结构及功能分析;(2)基于RNA测序的厌氧消化活性分析。在高通量测序技术出现之前,微生物群落结构分析主要采用分辨率很低的变性梯度凝胶电泳等分子指纹分析技术,通过切胶回收及传统Sanger测序分析,只能获得几十个序列。因此这些技术只能获得消化污泥中主要微生物种群,而无法获得整个污泥微生物组数据。后来随着高通量测序技术的发展,微生物群落(或微生物组)结构可以通过基于16S rRNA基因扩增子的高通量测序获得,通常一个样品能够获得几万到几十万个序列。此外,通过宏基因组及后续分箱分析,可以获得主要功能种群的基因组数据,并对整个消化污泥微生物组功能进行全面分析。另一方面,根据基因转录规律,通过对RNA进行高通量测序(RNA-Seq),并结合分箱分析所获得的主要功能种群基因组数据可以构建所有功能基因的转录全图,实现厌氧消化活性的系统分析;再通过结合实时荧光定量核酸扩增技术(qPCR)可以实现对少数目标基因转录水平的定量分析,最终实现关键功能基因的鉴定。下文将重点介绍基于高通量测序技术的16S rRNA基因测序及宏基因组、宏转录组分析技术与方法。

|
| 图 3 污泥厌氧消化微生物组分析流程图 Figure 3 Flow chart of microbiome analysis in anaerobic sludge digestion |
|
|
16S rRNA基因是编码原核微生物核糖体小亚基(16S rRNA)的1 500 bp基因。在微生物漫长进化过程中,由于16S rRNA基因碱基组成、核苷酸序列、DNA结构以及生物学功能等方面都表现出高度保守的特性,因而16S rRNA基因被称为“进化分子钟”来反映微生物种群间的系统进化及亲缘关系。基于16S rRNA基因序列测序的微生物组研究通常要经历以下6个步骤(图 4):待测生物样品收集、gDNA提取、聚合酶链式反应(Polymerase chain reaction,PCR)扩增、高通量测序、后续生物信息学数据处理和数据展示分析。

|
| 图 4 基于16S rRNA基因测序的污泥微生物组分析 Figure 4 Analysis of sludge microbiome based on 16S rRNA gene sequencing |
|
|
与传统低分辨率分子指纹分析技术相比,基于16S rRNA基因测序的污泥微生物组分析主要差别体现在PCR扩增、高通量测序及后续测序数据分析上。在PCR扩增阶段,为了能够将几百个样品混在一起进行后续分析,但又能够分清楚获得的序列来自于哪个样品,就需要利用带条码序列的引物(Barcoding primers)对各样品分别进行PCR扩增,再混合进行后续测序操作。在高通量测序平台选择上,目前测序技术主要经历了3代的发展:第1代以Sanger测序为代表,第2代高通量测序以Illumina测序平台为代表,第3代为单分子测序技术。目前使用的高通量测序平台主要基于Illumina平台所开发的MiSeq、HiSeq及NovaSeq等,主要因为第2代测序技术通量高、错误率低、成本低等特征[24-25]。
测序获得的海量数据在进行后续群落分析时需要进行数据前处理。例如,测序数据中常包括质量较低的DNA序列,需要通过生物信息学手段进行去除。原始高通量测序结果可以按照如下步骤进行处理:去除Adapters、Linkers、嵌合体和重复序列,删除小于某个长度的序列,去除测序质量值低的序列[26]。对于测序后数据的预处理,目前已经有研究人员开发了多个平台和软件,包括Trimmomatic[27]、UCHIME[28]等。随后,保留下来的有效序列可以通过USEARCH软件[29]、RDP Bayesian Classifier[30]、或者Uclust等聚类算法,按一定序列相似水平(一般为97%)将序列聚类成操作分类单元(Operational taxonomic units,OTUs)[31]。对获得的OTU代表序列,利用MOTHUR[32]、PyNAST[33]等序列对比软件进行数据库比对,包括SILVA、GreenGene或者RDP数据库[34]。进一步通过RDP朴素贝叶斯分类(Naive Bayesian Classifier)软件进行系统分类,得到每个样本所含的物种信息,继而进行后续生物信息统计分析。
获得上述信息后,可进一步进行微生物群落组成、系统发育树、α多样性、β多样性、分子网络及功能预测等分析。其中,系统发育树的建立可以通过ClusterX、MUSCLE等软件包进行多序列比对,利用MEGA、PHYLIP等程序计算不同序列差异性积分作为它们的差异性距离。进一步利用获得的各类群间进化距离构建系统发育树,然后通过距离标尺、分支长度、自展值等指标进行进化树评估。α多样性分析可以获得厌氧消化污泥微生物组结构的丰富度与多样性。基于OTU的α多样性研究可以依据Chao1丰富估计量(Chao1)进行分析,并进一步结合丰度覆盖的物种丰富度估计(Abundance coverage-based estimator of species richness,ACE)、Shannon-Weaver多样性指数(Shannon)、Simpson多样性指数(Simpson)进行分析。Chao1、ACE及Shannon指数与厌氧消化污泥微生物组的生物多样性呈正相关,而Simpson指数与生物多样性呈负相关。Chao1和ACE指数可直接用于计算微生物群落的物种总数或丰度,Shannon和Simpson指数可用于估算生物多样性[35],并且覆盖度越高,样品分析的可信度越高。例如,Sun等[36]通过污泥厌氧消化微生物α多样性分析,发现未进行污泥预处理的厌氧消化反应器内微生物多样性明显高于预处理污泥消化反应器。β多样性分析主要研究不同样品间的相似度和差异性,结果可以用主坐标分析(Principal co-ordinates analysis,PCoA)图及三元图表示。主坐标分析可以在R语言环境中利用GUniFrac和vegan等包计算各样品微生物组成的相似性距离,用ape包表征分析选择最能表示样本距离的坐标轴,得出各样品微生物组成的主坐标分析图。在主坐标分析图中,若2个样本之间的直线距离较近,则表示这2个样本的群落结构差异性较小。例如,Liu等利用β多样性分析揭示了厌氧生物反应器中长期生物强化(Bioaugmentation)和生物刺激(Biostimulation)处理会导致微生物组的不同改变[37]。三元图可以利用R语言中的ggtern包基于微生物物种进行相对丰度的三元图函数构建。样本中每个物种的相对丰度被定义为丰度本身与该物种在所有样本中总丰度的比例。每个物种在三元图中都有一个点,其大小可以根据相对丰度进行调整[35]。此外,还可以根据研究目的进行微生物分子网络分析确定环境因子与种群变化间的相关性,以及基于PICRUSt等软件包的群落功能分析[38-40]。Lu等基于分子网络分析鉴定了污泥厌氧消化反应器中盐度与污泥微生物组结构及功能间的联系[41]。
基于16S rRNA基因序列的微生物组分析具有快速、简单、样品制备和分析成本低、数据库容量大等特点,得到了广泛应用。但该方法也存在诸多缺陷。例如,同一个菌株基因组中可能存在多个16S rRNA基因拷贝[42-44],因此在统计丰度时会造成分析误差。Schmidt研究显示,在1 074类细菌及古菌中,只有20.2%的基因组存在单拷贝16S rRNA操纵子,而平均每个基因组存在3.94个16S rRNA操纵子,因此在计算群落中某个细菌丰度时,必须对每个基因组的16S rRNA基因拷贝数进行统计和综合计算,以便将测量到的基因拷贝数转换为实际的细胞数[45],这给数据分析带来不便。此外,仅16S rRNA基因序列信息可能不足以确定某个物种/菌株的系统发育关系,因为具有非常相似16S rRNA基因序列(如 > 99.5%相似性)的微生物基于DNA杂交结果实际上属于不同物种[46]。因此,复杂微生物组(例如厌氧消化污泥微生物组)的深入研究需要借助宏基因组分析。
2.2 基于宏基因组及宏转录组分析的微生物组研究宏基因组(Metagenome),也称为微生物环境基因组或元基因组(Microbial environmental genome),是由Handelsman等在1998年提出的新名词,定义为“The genomes of the total microbiota found in nature”,即以特定生境中的整个微生物群落作为研究对象,不需要对微生物进行分离培养,而是提取环境微生物总DNA进行研究。宏转录组兴起于宏基因组之后,从整体水平上研究某一特定环境、特定时期种群全部基因组转录情况以及转录调控规律。宏转录组分析可以揭示微生物在不同环境压力下的适应机制,探索环境与微生物之间的互作机理。对厌氧消化污泥微生物组进行宏基因组分析,不仅掌握消化污泥的群落结构(物种丰富度和分布),而且对群落功能(代谢)潜力会有更深层次的了解。此外,宏转录组通过提供各个基因的表达水平,结合宏基因组分析,可以进一步了解群落中每个种群相对转录活性,进而了解群落中不同种群功能基因的表达水平和调控关系。综合上述信息并结合各种群功能多样性,可以揭示群落中不同微生物种群的功能划分及群落组装模式。宏基因组及宏转录组分析流程如图 5所示。进行宏基因组测序分析时先对消化污泥微生物组样品进行DNA提取,与基于16S rRNA基因测序的微生物组研究不同,用于宏基因组分析的DNA样品无需进行PCR扩增。提取得到的厌氧消化污泥微生物组DNA后,将之进行随机打断并获取特定长度的短链DNA序列进行测序。对宏转录组分析来说,在对厌氧消化污泥微生物组进行RNA提取后,需立即利用反转录酶合成互补的双链cDNA文库,并用于后续的测序分析,以免RNA过度降解导致实验失败。以下将重点介绍宏基因组及宏转录组分析流程中的关键步骤。

|
| 图 5 污泥微生物组的宏基因组及宏转录组分析 Figure 5 Analysis of sludge microbiome based on metagenome and metatranscriptome |
|
|
(1) 宏基因组拼接
对于宏基因组测序数据,首先要对测序数据中的短片段进行拼接(Assemble),将其组装成长的重叠群(Contigs)再进行后续分析。目前已经有许多拼接方法被开发出来,而选择“最佳”拼接方法是一项具有挑战的任务,具体选择主要取决于研究目的。宏基因组从头组装(de novo assembly)在概念上类似于全基因组组装[47]。图论法(de Bruijn graph)是当前最被广泛使用的宏基因拼接方法。对于单个草图基因组(Draft genome)组装,通过将每个测序读长分解为一定长度k的重叠子序列来构建de Bruijn图。这组重叠的“k-mers”定义了de Bruijn图的顶点和边。拼接程序的主要任务是找到重建基因组的图形路径。但由于测序错误而产生的非基因组序列和重复序列将导致序列的错误拼接及碎片化,从而使得拼接这一过程变得复杂。另外,由于每个基因组成基因组的覆盖度取决于群落中每个基因组的丰度,如果总体测序深度不足将难以在图中形成连接,低丰度基因组可能最终碎片化。当前主要的拼接软件包括Meta-IDBA[48]、MetaVelvet[49]、metaSPAdes[50]、MEGAHIT[51]。根据宏基因组解释性评估(Critical assessment of metagenome interpretation,CAMI)报告[52],再基于其基准数据集进行测试,结果显示metaSPAdes及MEGAHIT软件是当前宏基因组拼接较好的软件。
(2) 重叠群分箱(Binning contigs)
宏基因组拼接高度分散,包含成千上万个重叠群,通常我们并不知道某个重叠群来自于哪个基因组,甚至不知道有多少基因组存在。重叠群分箱的目的主要是将重叠群归类形成物种,从而获得基因组草图(Draft genome)。监督式分箱法利用已经测序的基因组数据库来将重叠群标记到分类学特定级别。非监督式方法(聚类)则寻找数据中自然种群。监督和非监督方法都有两个主要元素:对重叠群和分箱之间相似性进行定义的度量,以及将这些相似性度量转换为赋值的算法。对于分类学分箱,针对已知基因组的重叠群同源性是一种潜在有用的方法,但大多数微生物物种尚未测序,因此大部分重建的基因组片段无法比对到参考基因组。而重叠群的聚类则很具吸引力,因为它不需要参考基因组。大多数重叠群聚类算法,例如MetaWat[53]和SCIMM[54]使用了各种物种组成指标,有时还与总覆盖度数据相结合。随着多样本宏基因组数据集的产生,研究人员已经意识到跨多个样本的重叠群覆盖提供了更强大的度量信号,能够将重叠群聚类在一起[55-56]。基本原则是来自同一基因组的重叠群将在每个宏基因组内具有相似的覆盖度,尽管基因组内GC含量的变化和细菌复制起点周围读长深度的增加可能会挑战这一假设[57]。例如最早的算法是基于二维可视化覆盖度信息和组成[55],需要人工输入来执行聚类,如扩展自组织映射[55]。而现在可以使用完全自动化的方法,例如CONCOCT[57]、Groop[58]和MetaBAT[59]更适用于大数据集。为了评估这些分类和组装的基因组质量,可以使用单拷贝基因分析工具,例如CheckM[60]利用常见单拷贝基因来评估基因组完整性和污染情况。经过分箱分析后的基因组草图则可用于后续分析。
(3) 基于重叠群序列的基因及群落组成分析
微生物群落分析中一个持久不变的主题是对微生物群落结构进行探索。较为常见的方法便是从宏基因组的拼接结果中恢复16S rRNA基因序列进行评估。例如,EMIRGE[61]从拼接结果中恢复16S rRNA基因全长序列用于群落组成分析。但由于重叠群高度分散化等特征,只有较少数完整的16S rRNA基因序列被恢复,因此造成群落组成多样性的低估。另外,CommunityM[62]则通过恢复16S rRNA基因全长序列或部分序列用于群落组成分析,对所获得的序列进行聚类形成OTU,并比对到原始的读长来统计丰度信息。然而,基于宏基因组测序与基于16S rRNA基因PCR扩增的群落组成上存在着差异,而Vanwonterghem等[62]认为这些差异主要是由PCR引物及扩增时的偏差所造成。
为了获得功能分类组成信息,可以使用生物信息学平台或BLAST等工具,利用数据库如SwissProt[63]、COG[64]、KEGG[64]、MG-RAST (SEED)[27]、GenDB[65]及IMG/M[66]对宏基因组数据进行功能注释。具体而言,BLAST主要通过与数据库进行比对的方法对宏基因组重叠群进行功能注释;MG-RAST服务器是一个基于SEED的环境,允许用户上传宏基因组以进行自动化功能注释分析;IMG/M为用户提供基因注释流程以及利用KEGG[62]、InterPro[67]、Pfam[68]等数据库为用户进行基因的比较分析。迄今为止,虽然已经开发了许多用于基因功能注释的生物信息学方法,但由于不均匀的序列组成、长度和测序错误,其中一部分如单基因组基因预测工具并不适合厌氧消化系统复杂宏基因组数据集[69]。不可否认的是,强大的基因注释工具(例如IMG/M+KEGG)仍然可以用于定位蛋白编码区域,这是宏基因组研究中的基本问题之一。例如Campanaro等[27]研究利用生物信息学工具(例如KEGG、COG及SEED)来描述参与厌氧消化过程的微生物物种间的功能性相互联系,并鉴定编码参与特定代谢途径功能酶的关键功能种群基因组。
(4) 基于基因组草图(Draft genome)的分析
重叠群分箱得到的基因组草图为获取单个物种的信息提供了便利。厌氧消化污泥微生物组极其复杂,虽然对其中大部分菌群的角色有一定认识,但仍然存在大量未知物种并行使着未知的功能。例如,从不同工程反应器中恢复的群落基因组提供了对候选分类门的基因组信息,包括TM7[56]和KSB3[70],分析发现上述2个物种分别与污水厌氧处理过程中丝状菌膨胀及对苯二甲酸降解相关[71]。对获得的基因组草图,可通过构建基因组系统发育树来确定其分类。目前用于构建系统发育树的方法很多,常用的软件包括PHYLIP[72]及RAxML[73]。其中PHYLIP运算速度较慢但精度较高,而RAxML在大数据集分析上有更优的运算速度。此外,对基因组草图中特定功能基因进行水平基因转移分析可以了解该物种在进化上的历史事件。基于基因组草图进行系统发育树的构建,不仅可以了解其分类地位及进化历史,还可以掌握其代谢通路并了解其生态学功能。进一步利用KEGG、COG及CAZy[74]等数据库对基因组草图进行注释,可以重建单个菌种代谢通路及微生物组中关键功能种群间的代谢网络。例如,Wang等结合厌氧微生物培养及宏基因组学分析,获得了还原脱氯微生物组中关键功能种群及其核心代谢网络(碳流与电子流网络)[75]。
(5) 宏转录组分析
宏转录组分析主要通过将宏转录组序列比对到经过宏基因组分箱的草图基因组上,例如利用Bowtie[76]软件进行比对,并且只允许一条读长分配到一条重叠群上。对于给定的基因,其丰度表征为完全落入该基因区域内的所有读长数目(即相应的基因完整地包括整个读长区间)。对于某一给定基因,其相对转录活性(Relative transcriptional activity,RTA)可通过以下算式获得:
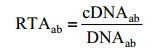
|
其中,cDNAab表示基因组b中基因a在该菌转录组中的相对丰度;DNAab表示基因组b中基因a在该菌基因组中的相对丰度。通过结合宏基因组与宏转录组数据分析可进一步获得关键功能基因信息。Wang等通过对厌氧还原脱卤培养物的宏基因组和宏转录组数据的分析,成功鉴定了这些培养物中厌氧还原脱卤关键功能基因[77]。同时,基于基因的相对转录活性,可进一步进行基因表达互作网络关系分析。
3 厌氧消化污泥微生物组分析案例研究者已经应用微生物组学分析方法成功鉴定了厌氧消化反应器中的优势种群[78-79],监测微生物组结构时空变化[80-81, 62],发现新甲烷菌[79, 82-84],探索产甲烷途径[85-87],分析厌氧消化对抗药基因(Antibiotic resistance genes,ARGs)影响[88-89],并阐明厌氧消化效率、微生物组与环境变量三者之间的关系[78, 85, 90-92]。上述研究中,充分了解厌氧消化过程中参与的微生物种群、相关功能基因及其互作关系尤为关键。这些信息对于设计、控制及优化厌氧消化过程极其重要[35]。近些年,在宏基因组学、宏转录组学及生物信息分析学快速发展的前提下,厌氧消化样品中关键功能种群、功能基因及其相互联系、相互作用的研究取得了许多进展[35]。例如,Campanaro等利用高通量鸟枪测序和一种新的分箱策略,对厌氧消化微生物组进行了深入的宏基因组分析,揭示了近100万个基因并获得106个完整微生物基因组[27]。确定了厌氧消化过程中关键代谢通路,如氨基酸发酵、脂肪酸降解、碳水化合物转化和乙酸盐氧化过程涉及的关键功能基因[27]。同时利用功能注释方法,结合KEGG数据库,确定了一种与甲烷单胞菌相关的新的未被培养的甲基营养型甲烷古菌[27]。利用相同的策略,该课题组分析了12家污水处理厂中/高温厌氧消化产甲烷的微生物组,研究人员利用Prodigal和FOAM软件分析了132个基因组在不同样本中的丰度及分布,从而确定了厌氧消化微生物组中的核心功能种群;此外,通过研究多个样本中的代谢通路与不同因子(如进料、温度、脂肪酸浓度及甲烷产量等)间的联系,揭示了高丰度核心功能种群间的主要联系,该研究为后来厌氧消化相关的研究提供了一种很有前景并获得广泛应用的分析策略[93]。
与传统厌氧消化微生物组的研究相比,Vanwoterghem等[62]以获取并分析功能种群基因组为中心,系统研究了实验室规模的污泥厌氧消化反应器中的微生物组(图 6),研究人员对纤维素转化为甲烷过程中涉及的功能种群进行了宏基因组分析,获得了101个关键功能种群的基因组,根据其潜在的底物利用情况,可以将这些基因组按照其功能性组合进行归类,通过恢复大多数种群的基因组,能够将各功能菌个体的代谢潜能与其相对丰度结合起来,并最终构建一个如图 6所示的清晰的代谢网络,该代谢网络显示出在水解和发酵过程中存在较高水平的功能冗余,随着时间推移,涉及的主要功能种群丰度在变化,但同时保持着厌氧消化整体功能的完整性;同时,由于Bact_03、Actino_02等具有Methylmalonyl-CoA代谢通路(与丙酸盐产生相关)的菌群在反应器T1运行阶段相对丰度较高,通过代谢网络与相关影响因子的关联分析,很好地解释了实验中丙酸盐累积的现象;基因组解析还可以计算出每个功能种群的相对丰度比例,计算结果突出了具有功能多样性、灵活性并保持良好平衡的各功能种群如何协调并共同完成复杂、多步骤的污泥厌氧消化过程。同时,根据相关代谢特征,划分不同功能种群的角色,并能确定一些罕见的未被培养的微生物种群在污泥厌氧消化过程中的功能。

剩余(活性)污泥厌氧消化是今后污泥减量化、稳定化处理处置的关键环节之一。由于厌氧消化污泥的复杂微生物组,污泥厌氧消化也因此成为整个污泥处理、处置流程的难点所在。环境微生物组学研究工具的快速发展为该难点问题的解决提供了有效的技术手段,未来在厌氧消化污泥微生物组研究及应用中亟待解决以下几个问题,这些问题的解决将促进厌氧消化污泥微生物组的研究及应用。
(1) 污泥微生物组研究需要自己的生物信息学分析流程及数据库。基于高通量测序的宏基因组及宏转录组分析需要依赖健全的参照数据库及合适的近似算法。当前宏基因组及宏转录组数据库及分析软件或程序主要针对生命科学(如肠道微生物组)、农业与食品发酵科学及海洋科学等学科进行开发。将这些数据库及程序直接用于厌氧消化污泥微生物组的研究,需要投入更多计算力和其它资源去比对不同方法产生的分析结果,并且很容易产生误差。
(2) 清晰界定厌氧消化污泥微生物组中关键功能种群、冗余种群及其它干扰种群。污泥厌氧消化反应器与普通消化反应器不同,进料污泥中含有大量微生物细胞,而且剩余活性污泥中含有大量兼性及少量厌氧微生物种群,这些种群进入污泥厌氧消化反应器后,并不会作为底物被消化功能种群降解,而是继续在厌氧消化污泥中存活并进行代谢,因此在进行厌氧消化污泥微生物组分析时会产生干扰,影响到消化污泥微生物组组成及功能的分析,更会影响到种群间互作关系的研究。因此,如何准确区分厌氧消化污泥微生物组中关键功能种群、冗余种群及其它干扰种群,仍然是厌氧消化污泥微生物研究的难点问题。
(3) 消化污泥中微生物资源开发、利用:厌氧消化污泥中存在大量不可培养或未被培养的微生物种群。目前这些微生物种群还未被分类,它们的生理生化特征也未被鉴定。因此这些微生物种群被称为污泥消化反应器中的微生物暗物质(Microbial dark matter)。这些微生物暗物质的存在成为消化污泥微生物组研究的障碍,但同时也预示着厌氧消化污泥中丰富的微生物资源有待开发。而这部分的研究工作高度依赖宏基因组及传统微生物培养技术的有机结合:通过宏基因组分析,首先获得这些微生物暗物质的代谢潜力并据此确定选择性培养条件,再结合传统微生物培养技术进行富集培养、分离及表征。
| [1] |
Li XS, Ma HZ, Wang QH, et al. Isolation, identification of sludge-lysing strain and its utilization in thermophilic aerobic digestion for waste activated sludge[J]. Bioresource Technology, 2009, 100(9): 2475-2481. DOI:10.1016/j.biortech.2008.12.019 |
| [2] |
Yang G, Zhang GM, Wang HC. Current state of sludge production, management, treatment and disposal in China[J]. Water Research, 2015, 78: 60-73. DOI:10.1016/j.watres.2015.04.002 |
| [3] |
Wei YS, van Houten RT, Borger AR, et al. Minimization of excess sludge production for biological wastewater treatment[J]. Water Research, 2003, 37(18): 4453-4467. DOI:10.1016/S0043-1354(03)00441-X |
| [4] |
Ju F, Lau F, Zhang T. Linking microbial community, environmental variables, and methanogenesis in anaerobic biogas digesters of chemically enhanced primary treatment sludge[J]. Environmental Science & Technology, 2017, 51(7): 3982-3992. |
| [5] |
Hassa J, Maus I, Off S, et al. Metagenome, metatranscriptome, and metaproteome approaches unraveled compositions and functional relationships of microbial communities residing in biogas plants[J]. Applied Microbiology and Biotechnology, 2018, 102(12): 5045-5063. DOI:10.1007/s00253-018-8976-7 |
| [6] |
Keegan KP, Glass EM, Meyer F. MG-RAST, a metagenomics service for analysis of microbial community structure and function[A]//Martin F, Uroz S. Microbial Environmental Genomics (MEG)[M]. New York, NY: Humana Press, 2016, 1399: 207-233
|
| [7] |
Laspidou CS, Rittmann BE. A unified theory for extracellular polymeric substances, soluble microbial products, and active and inert biomass[J]. Water Research, 2002, 36(11): 2711-2720. DOI:10.1016/S0043-1354(01)00413-4 |
| [8] |
Kelessidis A, Stasinakis AS. Comparative study of the methods used for treatment and final disposal of sewage sludge in European countries[J]. Waste Management, 2012, 32(6): 1186-1195. DOI:10.1016/j.wasman.2012.01.012 |
| [9] |
Subramanian SB, Yan S, Tyagi RD, et al. Extracellular polymeric substances (EPS) producing bacterial strains of municipal wastewater sludge: isolation, molecular identification, EPS characterization and performance for sludge settling and dewatering[J]. Water Research, 2010, 44(7): 2253-2266. DOI:10.1016/j.watres.2009.12.046 |
| [10] |
Gonzalez A, Hendriks ATWM, van Lier JB, et al. Pre-treatments to enhance the biodegradability of waste activated sludge: Elucidating the rate limiting step[J]. Biotechnology Advances, 2018, 36(5): 1434-1469. DOI:10.1016/j.biotechadv.2018.06.001 |
| [11] |
Seviour RJ, Nielsen PH. Microbial Ecology of Activated Sludge[M]. London: IWA Publishing, 2010: 95-125.
|
| [12] |
Sheng GP, Yu HQ, Li XY. Extracellular polymeric substances (EPS) of microbial aggregates in biological wastewater treatment systems: a review[J]. Biotechnology Advances, 2010, 28(6): 882-894. DOI:10.1016/j.biotechadv.2010.08.001 |
| [13] |
Liu XM, Sheng GP, Luo HW, et al. Contribution of extracellular polymeric substances (EPS) to the sludge aggregation[J]. Environmental Science & Technology, 2010, 44(11): 4355-4360. |
| [14] |
Zhen GY, Lu XQ, Kato H, et al. Overview of pretreatment strategies for enhancing sewage sludge disintegration and subsequent anaerobic digestion: current advances, full-scale application and future perspectives[J]. Renewable and Sustainable Energy Reviews, 2017, 69: 559-577. DOI:10.1016/j.rser.2016.11.187 |
| [15] |
Schimel KA. Biogas plasticization coupled anaerobic digestion: the anaerobic pump stoichiometry[J]. Applied Biochemistry and Biotechnology, 2014, 172(4): 2227-2252. DOI:10.1007/s12010-013-0558-7 |
| [16] |
Zeikus JG. Anaerobic Digestion[M]. London: Applied Science Publisher, 1980: 61-87.
|
| [17] |
Jain S, Jain S, Wolf IT, et al. A comprehensive review on operating parameters and different pretreatment methodologies for anaerobic digestion of municipal solid waste[J]. Renewable and Sustainable Energy Reviews, 2015, 52: 142-154. DOI:10.1016/j.rser.2015.07.091 |
| [18] |
Guo JH, Peng YZ, Ni BJ, et al. Dissecting microbial community structure and methane-producing pathways of a full-scale anaerobic reactor digesting activated sludge from wastewater treatment by metagenomic sequencing[J]. Microbial Cell Factories, 2015, 14: 33. DOI:10.1186/s12934-015-0218-4 |
| [19] |
Cai MW, Wilkins D, Chen JP, et al. Metagenomic reconstruction of key anaerobic digestion pathways in municipal sludge and industrial wastewater biogas-producing systems[J]. Frontiers in Microbiology, 2016, 7: 778. |
| [20] |
Foladori P, Velho VF, Costa RHR, et al. Concerning the role of cell lysis-cryptic growth in anaerobic side-stream reactors: the single-cell analysis of viable, dead and lysed bacteria[J]. Water Research, 2015, 74: 132-142. DOI:10.1016/j.watres.2015.01.042 |
| [21] |
Wett B, Phothilangka P, Eladawy A. Systematic comparison of mechanical and thermal sludge disintegration technologies[J]. Waste Management, 2010, 30(6): 1057-1062. DOI:10.1016/j.wasman.2009.12.011 |
| [22] |
Peng LY, Appels L, Su HJ. Combining microwave irradiation with sodium citrate addition improves the pre-treatment on anaerobic digestion of excess sewage sludge[J]. Journal of Environmental Management, 2018, 213: 271-278. |
| [23] |
Nazari L, Yuan Z, Santoro D, et al. Low-temperature thermal pre-treatment of municipal wastewater sludge: Process optimization and effects on solubilization and anaerobic degradation[J]. Water Research, 2017, 113: 111-123. DOI:10.1016/j.watres.2016.11.055 |
| [24] |
Heather JM, Chain B. The sequence of sequencers: the history of sequencing DNA[J]. Genomics, 2016, 107(1): 1-8. |
| [25] |
White Ⅲ RA, Callister SJ, Moore RJ, et al. The past, present and future of microbiome analyses[J]. Nature Protocols, 2016, 11(11): 2049-2053. DOI:10.1038/nprot.2016.148 |
| [26] |
Li S, Chou HH. LUCY2: an interactive DNA sequence quality trimming and vector removal tool[J]. Bioinformatics, 2004, 20(16): 2865-2866. DOI:10.1093/bioinformatics/bth302 |
| [27] |
Campanaro S, Treu L, Kougias PG, et al. Metagenomic analysis and functional characterization of the biogas microbiome using high throughput shotgun sequencing and a novel binning strategy[J]. Biotechnology for Biofuels, 2016, 9: 26. DOI:10.1186/s13068-016-0441-1 |
| [28] |
Maspolim Y, Zhou Y, Guo CH, et al. Determination of the archaeal and bacterial communities in two-phase and single-stage anaerobic systems by 454 pyrosequencing[J]. Journal of Environmental Sciences, 2015, 36: 121-129. DOI:10.1016/j.jes.2015.02.017 |
| [29] |
Wilkins D, Rao S, Lu XY, et al. Effects of sludge inoculum and organic feedstock on active microbial communities and methane yield during anaerobic digestion[J]. Frontiers in Microbiology, 2015, 6: 1114. |
| [30] |
Cardinali-Rezende J, Rojas-Ojeda P, Nascimento AMA, et al. Proteolytic bacterial dominance in a full-scale municipal solid waste anaerobic reactor assessed by 454 pyrosequencing technology[J]. Chemosphere, 2016, 146: 519-525. DOI:10.1016/j.chemosphere.2015.12.003 |
| [31] |
Cole JR, Chai B, Farris RJ, et al. The Ribosomal Database Project (RDP-Ⅱ): sequences and tools for high-throughput rRNA analysis[J]. Nucleic Acids Research, 2005, 33(S1): D294-D296. |
| [32] |
Su HJ, Liu L, Wang SJ, et al. Semi-continuous anaerobic digestion for biogas production: influence of ammonium acetate supplement and structure of the microbial community[J]. Biotechnology for Biofuels, 2015, 8: 13. DOI:10.1186/s13068-015-0197-z |
| [33] |
Oosterkamp MJ, Méndez-García C, Kim CH, et al. Lignocellulose-derived thin stillage composition and efficient biological treatment with a high-rate hybrid anaerobic bioreactor system[J]. Biotechnology for Biofuels, 2016, 9: 120. DOI:10.1186/s13068-016-0532-z |
| [34] |
Wang Q, Garrity GM, Tiedje JM, et al. Naive bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy[J]. Applied and Environmental Microbiology, 2007, 73(16): 5261-5267. DOI:10.1128/AEM.00062-07 |
| [35] |
Zhang L, Loh KC, Lim JW, et al. Bioinformatics analysis of metagenomics data of biogas-producing microbial communities in anaerobic digesters: a review[J]. Renewable and Sustainable Energy Reviews, 2019, 100: 110-126. DOI:10.1016/j.rser.2018.10.021 |
| [36] |
Sun R, Xing DF, Jia JN, et al. Methane production and microbial community structure for alkaline pretreated waste activated sludge[J]. Bioresource Technology, 2014, 169: 496-501. DOI:10.1016/j.biortech.2014.07.032 |
| [37] |
Liu MM, Wang SQ, Nobu MK, et al. Impacts of biostimulation and bioaugmentation on the performance and microbial ecology in methanogenic reactors treating purified terephthalic acid wastewater[J]. Water Research, 2017, 122: 308-316. DOI:10.1016/j.watres.2017.06.008 |
| [38] |
Okuda S, Tsuchiya Y, Kiriyama C, et al. Virtual metagenome reconstruction from 16S rRNA gene sequences[J]. Nature Communications, 2012, 3: 1203. DOI:10.1038/ncomms2203 |
| [39] |
Aßhauer KP, Wemheuer B, Daniel R, et al. Tax4Fun: predicting functional profiles from metagenomic 16S rRNA data[J]. Bioinformatics, 2015, 31(17): 2882-2884. DOI:10.1093/bioinformatics/btv287 |
| [40] |
Scholz M, Ward DV, Pasolli E, et al. Strain-level microbial epidemiology and population genomics from shotgun metagenomics[J]. Nature Methods, 2016, 13(5): 435-438. DOI:10.1038/nmeth.3802 |
| [41] |
Lu QH, Yu ZH, Yu SN, et al. Organic matter rather than salinity as a predominant feature governing performance and microbiome in methanogenic sludge digesters[J]. Journal of Hazardous Materials, 2019. DOI:10.1016/j.jhazmat.2019.05.075 |
| [42] |
Yamamoto S, Harayama S. PCR amplification and direct sequencing of gyrB genes with universal primers and their application to the detection and taxonomic analysis of Pseudomonas putida strains[J]. Applied and Environmental Microbiology, 1995, 61(3): 1104-1109. |
| [43] |
Klappenbach JA, Saxman PR, Cole JR, et al. rrndb: the ribosomal RNA operon copy number database[J]. Nucleic Acids Research, 2001, 29(1): 181-184. DOI:10.1093/nar/29.1.181 |
| [44] |
Acinas SG, Marcelino LA, Klepac-Ceraj V, et al. Divergence and redundancy of 16S rRNA sequences in genomes with multiple rrn operons[J]. Journal of Bacteriology, 2004, 186(9): 2629-2635. DOI:10.1128/JB.186.9.2629-2635.2004 |
| [45] |
Schmidt TM. rrnDB: documenting the number of rRNA and tRNA genes in bacteria and archaea[J]. Nucleic Acids Research, 2009, 37(Database): D489-D493. DOI:10.1093/nar/gkn689 |
| [46] |
Fox GE, Wisotzkey JD, Jurtshuk Jr P. How close is close: 16S rRNA sequence identity may not be sufficient to guarantee species identity[J]. International Journal of Systematic Bacteriology, 1992, 42(1): 166-170. DOI:10.1099/00207713-42-1-166 |
| [47] |
Simpson JT, Pop M. The theory and practice of genome sequence assembly[J]. Annual Review of Genomics and Human Genetics, 2015, 16: 153-172. DOI:10.1146/annurev-genom-090314-050032 |
| [48] |
Peng Y, Leung HCM, Yiu SM, et al. Meta-IDBA: a de novo assembler for metagenomic data[J]. Bioinformatics, 2011, 27(13): i94-i101. DOI:10.1093/bioinformatics/btr216 |
| [49] |
Namiki T, Hachiya T, Tanaka H, et al. MetaVelvet: an extension of Velvet assembler to de novo metagenome assembly from short sequence reads[J]. Nucleic Acids Research, 2012, 40(20): e155. DOI:10.1093/nar/gks678 |
| [50] |
Bankevich A, Nurk S, Antipov D, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing[J]. Journal of Computational Biology, 2012, 19(5): 455-477. DOI:10.1089/cmb.2012.0021 |
| [51] |
Li DH, Liu CM, Luo RB, et al. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph[J]. Bioinformatics, 2015, 31(10): 1674-1676. DOI:10.1093/bioinformatics/btv033 |
| [52] |
Sczyrba A, Hofmann P, Belmann P, et al. Critical assessment of metagenome interpretation-a benchmark of metagenomics software[J]. Nature Methods, 2017, 14(11): 1063-1071. DOI:10.1038/nmeth.4458 |
| [53] |
Strous M, Kraft B, Bisdorf R, et al. The binning of metagenomic contigs for microbial physiology of mixed cultures[J]. Frontiers in Microbiology, 2012, 3: 410. |
| [54] |
Kelley DR, Salzberg SL. Clustering metagenomic sequences with interpolated Markov models[J]. BMC Bioinformatics, 2010, 11: 544. DOI:10.1186/1471-2105-11-544 |
| [55] |
Sharon I, Morowitz MJ, Thomas BC, et al. Time series community genomics analysis reveals rapid shifts in bacterial species, strains, and phage during infant gut colonization[J]. Genome Research, 2013, 23(1): 111-120. DOI:10.1101/gr.142315.112 |
| [56] |
Albertsen M, Hugenholtz P, Skarshewski A, et al. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes[J]. Nature Biotechnology, 2013, 31(6): 533-538. DOI:10.1038/nbt.2579 |
| [57] |
Korem T, Zeevi D, Suez J, et al. Growth dynamics of gut microbiota in health and disease inferred from single metagenomic samples[J]. Science, 2015, 349(6252): 1101-1106. DOI:10.1126/science.aac4812 |
| [58] |
Imelfort M, Parks D, Woodcroft BJ, et al. GroopM: an automated tool for the recovery of population genomes from related metagenomes[J]. PeerJ, 2014, 2: e603. DOI:10.7717/peerj.603 |
| [59] |
Kang DD, Froula J, Egan R, et al. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities[J]. PeerJ, 2015, 3: e1165. DOI:10.7717/peerj.1165 |
| [60] |
Parks DH, Imelfort M, Skennerton CT, et al. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes[J]. Genome Research, 2015, 25(7): 1043-1055. DOI:10.1101/gr.186072.114 |
| [61] |
Miller CS, Baker BJ, Thomas BC, et al. EMIRGE: reconstruction of full-length ribosomal genes from microbial community short read sequencing data[J]. Genome Biology, 2011, 12(5): R44. DOI:10.1186/gb-2011-12-5-r44 |
| [62] |
Vanwonterghem I, Jensen PD, Rabaey K, et al. Genome-centric resolution of microbial diversity, metabolism and interactions in anaerobic digestion[J]. Environmental Microbiology, 2016, 18(9): 3144-3158. DOI:10.1111/1462-2920.13382 |
| [63] |
Boeckmann B, Bairoch A, Apweiler R, et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003[J]. Nucleic Acids Research, 2003, 31(1): 365-370. DOI:10.1093/nar/gkg095 |
| [64] |
Schlüter A, Bekel T, Diaz NN, et al. The metagenome of a biogas-producing microbial community of a production-scale biogas plant fermenter analysed by the 454-pyrosequencing technology[J]. Journal of Biotechnology, 2008, 136(1/2): 77-90. |
| [65] |
Meyer F, Goesmann A, McHardy AC, et al. GenDB-an open source genome annotation system for prokaryote genomes[J]. Nucleic Acids Research, 2003, 31(8): 2187-2195. DOI:10.1093/nar/gkg312 |
| [66] |
Markowitz VM, Chen IMA, Chu K, et al. IMG/M 4 version of the integrated metagenome comparative analysis system[J]. Nucleic Acids Research, 2014, 42(D1): D568-D573. DOI:10.1093/nar/gkt919 |
| [67] |
Hunter S, Apweiler R, Attwood TK, et al. InterPro: the integrative protein signature database[J]. Nucleic Acids Research, 2009, 37(S1): D211-D215. |
| [68] |
Finn RD, Bateman A, Clements J, et al. Pfam: the protein families database[J]. Nucleic Acids Research, 2014, 42(D1): D222-D230. DOI:10.1093/nar/gkt1223 |
| [69] |
Jünemann S, Kleinbölting N, Jaenicke S, et al. Bioinformatics for NGS-based metagenomics and the application to biogas research[J]. Journal of Biotechnology, 2017, 261: 10-23. DOI:10.1016/j.jbiotec.2017.08.012 |
| [70] |
Sekiguchi Y, Ohashi A, Parks DH, et al. First genomic insights into members of a candidate bacterial phylum responsible for wastewater bulking[J]. PeerJ, 2015, 3: e740. DOI:10.7717/peerj.740 |
| [71] |
Nobu MK, Narihiro T, Rinke C, et al. Microbial dark matter ecogenomics reveals complex synergistic networks in a methanogenic bioreactor[J]. The ISME Journal, 2015, 9(8): 1710-1722. DOI:10.1038/ismej.2014.256 |
| [72] |
Retief JD. Phylogenetic analysis using PHYLIP[A]//Misener S, Krawetz SA. Bioinformatics Methods and Protocols[M]. Totowa, NJ: Humana Press, 1999, 132: 243-258
|
| [73] |
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies[J]. Bioinformatics, 2014, 30(9): 1312-1313. DOI:10.1093/bioinformatics/btu033 |
| [74] |
Lombard V, Golaconda Ramulu H, Drula E, et al. The carbohydrate-active enzymes database (CAZy) in 2013[J]. Nucleic Acids Research, 2014, 42(D1): D490-D495. DOI:10.1093/nar/gkt1178 |
| [75] |
Wang SQ, Chen C, Zhao SY, et al. Microbial synergistic interactions for reductive dechlorination of polychlorinated biphenyls[J]. Science of the Total Environment, 2019, 666: 368-376. DOI:10.1016/j.scitotenv.2019.02.283 |
| [76] |
Langmead B. Aligning short sequencing reads with bowtie[J]. Current Protocols in Bioinformatics, 2010, 32(1): 11.7.1-11.7.14. |
| [77] |
Wang SQ, Chng KR, Wilm A, et al. Genomic characterization of three unique Dehalococcoides that respire on persistent polychlorinated biphenyls[J]. Proceedings of the National Academy of Sciences of the United States of America, 2014, 111(33): 12103-12108. DOI:10.1073/pnas.1404845111 |
| [78] |
Luo G, Fotidis IA, Angelidaki I. Comparative analysis of taxonomic, functional, and metabolic patterns of microbiomes from 14 full-scale biogas reactors by metagenomic sequencing and radioisotopic analysis[J]. Biotechnology for Biofuels, 2016, 9: 51. DOI:10.1186/s13068-016-0465-6 |
| [79] |
Chouari R, Le Paslier D, Daegelen P, et al. Novel predominant archaeal and bacterial groups revealed by molecular analysis of an anaerobic sludge digester[J]. Environmental Microbiology, 2005, 7(8): 1104-1115. DOI:10.1111/j.1462-2920.2005.00795.x |
| [80] |
Vanwonterghem I, Jensen PD, Dennis PG, et al. Deterministic processes guide long-term synchronised population dynamics in replicate anaerobic digesters[J]. The ISME Journal, 2014, 8(10): 2015-2028. DOI:10.1038/ismej.2014.50 |
| [81] |
Peces M, Astals S, Jensen PD, et al. Deterministic mechanisms define the long-term anaerobic digestion microbiome and its functionality regardless of the initial microbial community[J]. Water Research, 2018, 141: 366-376. DOI:10.1016/j.watres.2018.05.028 |
| [82] |
Nobu MK, Narihiro T, Kuroda K, et al. Chasing the elusive Euryarchaeota class WSA2: genomes reveal a uniquely fastidious methyl-reducing methanogen[J]. The ISME Journal, 2016, 10(10): 2478-2487. DOI:10.1038/ismej.2016.33 |
| [83] |
Vanwonterghem I, Evans PN, Parks DH, et al. Methylotrophic methanogenesis discovered in the archaeal phylum Verstraetearchaeota[J]. Nature Microbiology, 2016, 1(12): 16170. DOI:10.1038/nmicrobiol.2016.170 |
| [84] |
McIlroy SJ, Kirkegaard RH, Dueholm MS, et al. Culture-independent analyses reveal novel Anaerolineaceae as abundant primary fermenters in anaerobic digesters treating waste activated sludge[J]. Frontiers in Microbiology, 2017, 8: 1134. DOI:10.3389/fmicb.2017.01134 |
| [85] |
Li A, Chu YN, Wang XM, et al. A pyrosequencing-based metagenomic study of methane-producing microbial community in solid-state biogas reactor[J]. Biotechnology for Biofuels, 2013, 6(1): 3. |
| [86] |
Wong MT, Zhang D, Li J, et al. Towards a metagenomic understanding on enhanced biomethane production from waste activated sludge after pH 10 pretreatment[J]. Biotechnology for Biofuels, 2013, 6(1): 38. |
| [87] |
Tian Z, Zhang Y, Yu B, et al. Changes of resistome, mobilome and potential hosts of antibiotic resistance genes during the transformation of anaerobic digestion from mesophilic to thermophilic[J]. Water Research, 2016, 98: 261-269. DOI:10.1016/j.watres.2016.04.031 |
| [88] |
Jang HM, Shin J, Choi S, et al. Fate of antibiotic resistance genes in mesophilic and thermophilic anaerobic digestion of chemically enhanced primary treatment (CEPT) sludge[J]. Bioresource Technology, 2017, 244: 433-444. DOI:10.1016/j.biortech.2017.07.153 |
| [89] |
Koo T, Shin SG, Lee J, et al. Identifying methanogen community structures and their correlations with performance parameters in four full-scale anaerobic sludge digesters[J]. Bioresource Technology, 2017, 228: 368-373. DOI:10.1016/j.biortech.2016.12.118 |
| [90] |
Hidaka T, Tsushima I, Tsumori J. Comparative analyses of microbial structures and gene copy numbers in the anaerobic digestion of various types of sewage sludge[J]. Bioresource Technology, 2018, 253: 315-322. DOI:10.1016/j.biortech.2017.12.097 |
| [91] |
De Vrieze J, Saunders AM, He Y, et al. Ammonia and temperature determine potential clustering in the anaerobic digestion microbiome[J]. Water Research, 2015, 75: 312-323. DOI:10.1016/j.watres.2015.02.025 |
| [92] |
Pervin HM, Dennis PG, Lim HJ, et al. Drivers of microbial community composition in mesophilic and thermophilic temperature-phased anaerobic digestion pre-treatment reactors[J]. Water Research, 2013, 47(19): 7098-7108. DOI:10.1016/j.watres.2013.07.053 |
| [93] |
Campanaro S, Treu L, Kougias PG, et al. Metagenomic binning reveals the functional roles of core abundant microorganisms in twelve full-scale biogas plants[J]. Water Research, 2018, 140: 123-134. DOI:10.1016/j.watres.2018.04.043 |