 2019, Vol. 46
2019, Vol. 46扩展功能
文章信息
- 吴麒, 陈颖, 邱凯瑞, 罗倩倩
- WU Qi, CHEN Ying, QIU Kai-Rui, LUO Qian-Qian
- 产甲烷条件下岩溶湿地沉积物中古菌群落的变化规律
- Characterization of archaeal communities in a karst wetland under methanogenic conditions
- 微生物学通报, 2019, 46(12): 3193-3204
- Microbiology China, 2019, 46(12): 3193-3204
- DOI: 10.13344/j.microbiol.china.190049
-
文章历史
- 收稿日期: 2019-01-17
- 接受日期: 2019-05-28
- 网络首发日期: 2019-05-31

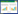




岩溶环境约占全球陆地面积的13.8%[1],全球约有1/4人口的饮用水部分或完全来自于岩溶环境的水资源[2]。岩溶环境分布于地表和地下,由溶蚀性的水对可溶性岩石破坏和改造形成。岩溶作用的主要机制有两种:二氧化碳溶解于水后形成的碳酸按照反应式(1)和(2)对碳酸盐岩石的溶蚀是一类常见的岩溶过程,反应产生的钙离子和碳酸氢根离子使岩溶环境的水体通常呈现富钙偏碱的特征;在含硫的岩溶环境,水中的硫酸也具有岩溶作用。
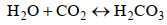
|
(1) |
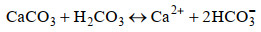
|
(2) |
岩溶作用造就了岩溶环境独特的水循环系统,通常包括地表的湿地、湖泊以及广阔的地下水系等,具有水量丰富而不均、多重水介质并存、流态变化复杂以及伏流和明流交替出现等特征[3]。水流在改造岩溶地貌的同时连接了地表和地下的岩溶生态系统,也影响着其中的微生物群落组成和功能。对一部分岩溶环境而言,能够获得较多来自地表的有机物输入,其中的微生物以有机营养型为主;一部分相对封闭的岩溶环境很少或没有从地表环境获得有机物,处于寡营养状态;还有一部分岩溶环境富含硫化物、氨或甲烷等物质,栖息了许多化能营养型微生物[4]。
在缺少氧气、硝酸盐和硫酸盐等各类电子受体的条件下,产甲烷作用是有机物降解的最后环节。甲烷是一种清洁能源,也是一种温室效应气体,因此,产甲烷作用对全球气候、碳循环、生态环境以及社会发展都具有重要的影响。产甲烷作用主要由古菌介导完成,全球每年排放到大气的甲烷中,约有70%以上来自微生物的产甲烷活动[5]。目前已知的产甲烷菌主要属于广古菌门中的7个目,包括甲烷微菌目(Methanomicrobiales)、甲烷杆菌目(Methanobacteriales)和甲烷八叠球菌目(Methanosarcinales)等,能将有机物厌氧降解产生的氢气和二氧化碳、乙酸、甲基类物质等小分子物质转变成甲烷,并通过这种方式获得生长所需的能量和碳源。氢型产甲烷和乙酸型产甲烷是主要的产甲烷方式,分别贡献了每年微生物产甲烷总量的约33%和67%,其他底物型产甲烷方式的贡献估计小于5%[6-7]。另外,近年来也有研究表明,深古菌门(Bathyarchaeota)和韦斯特(Verstraetearchaeota)古菌门的部分成员中也具有产甲烷菌代谢途径相关的基因,推测这些古菌可能也具有产甲烷的能力[8-9]。
岩溶环境有着独特的地质构造和水文特征,也蕴含着许多新型产甲烷菌。近年来人们从岩溶环境中发现了许多新型的产甲烷菌,如澳大利亚Wellington Caves的两处岩溶洞穴土壤中发现的产甲烷菌约有50%−70%属于尚未分类的类型;罗马尼亚Movile Cave分离出了产甲烷菌新种Methanobacterium movilense和Methanosarcina spelaei[10-11]。
与其他环境中的产甲烷菌类似,岩溶环境产甲烷菌的分布也受到相似的环境因子影响。产甲烷菌栖息在严格厌氧的环境,在洞穴深层湿润缺氧的土壤[12],以及岩溶湖泊的底层水体和沉积物中[13-14],广古菌门的产甲烷菌都是主要的古菌类群;在富含硫酸盐和硝酸盐等电子受体的环境,硫酸盐还原菌和硝酸盐还原菌可能会与产甲烷菌竞争乙酸和氢气等物质,产甲烷菌通常会受到这些竞争性电子受体的抑制,目前许多岩溶环境古菌的研究都是围绕富含硫酸盐和硝酸盐的岩溶环境,在这类岩溶环境中产甲烷菌的数量和活性通常都很低或未检测到[15-19];另外,水体的富营养化会增加产甲烷菌的数量和活性[20],但在岩溶环境中相关研究未见报道。
岩溶环境蕴藏着丰富的水资源,包括产甲烷菌在内的古菌是有机质降解的重要贡献者,在水资源匮乏和环境污染日趋严峻的背景下,研究岩溶环境古菌具有重要的意义。会仙岩溶湿地是我国中低纬度低海拔地区最具代表性的自然湿地之一[21],地处桂林市临桂区境内,位于石灰岩峰丛洼地与峰林平原的过渡区,其中包含运河、岩溶湖泊和沼泽,经由地上和地下水系相互交联,对净化漓江水质和保护漓江流域生物多样性具有重要作用。我们之前对会仙岩溶湿地产甲烷菌的原位研究显示,其沉积物中硫酸盐和硝酸盐的浓度都较低,有机物的含量比较高,且蕴涵着较为丰富和多样的产甲烷菌[22],是研究岩溶环境产甲烷菌理想的材料。
本文对会仙岩溶湿地的沉积物样品分别进行了以氢气为唯一能量来源和以乙酸钠为唯一能量来源的厌氧富集培养,构建了古菌16S rRNA基因全长文库和甲基辅酶M还原酶基因mcrA文库。mcrA基因是产甲烷菌的特征基因,用于研究已知产甲烷古菌的群落组成,古菌16S rRNA基因文库则用于揭示产甲烷条件下直接或间接利用产甲烷底物的古菌。同时,本文还系统地比较和分析了两种产甲烷富集培养条件下产甲烷菌和其他古菌的群落构成和文库中序列的归属,以期有助于揭示会仙岩溶湿地沉积物中氢型产甲烷菌和乙酸型产甲烷菌的主要类群,探究岩溶沉积物中古菌群落结构对产甲烷条件的响应,为开发和保护岩溶湿地微生物资源提供理论基础和技术支持。
1 材料与方法 1.1 主要试剂和仪器土壤DNA提取试剂盒、胶回收试剂盒,Omega公司;PCR扩增试剂盒、pMD18-T载体克隆试剂盒,北京宝日生物技术有限公司。
活塞式柱状沉积物采样器,上虞音花仪器厂;气相色谱仪,Agilent Technologies公司;气体采样器,Hamilton公司;气体采样袋,上海申源公司;厌氧培养瓶、顶空型试剂瓶盖,Schott公司;培养瓶丁基胶塞,Glasgerätebau Ochs公司;PCR扩增仪,Bio-Rad公司。
1.2 培养基富集培养用培养基改良自ATCC 1340 MS培养基[23],原培养基中的酵母膏、蛋白胨和甲酸盐被排除,硫酸盐成分由对应的盐酸盐替代。
1.3 样品采集和地化参数的测定样品于2016年9月24日采集自中国地质科学院会仙岩溶生态与水生态研究基地,研究基地位于会仙岩溶湿地中央岩溶盆地地势低洼区域,距桂林市区西南约30公里(N25°06′01.8″,E110°13′55.1″)。当日气温30 ℃,池底水温26 ℃。柱状沉积物通过活塞式柱状沉积物采样器采集,样品采集后选取其中15 cm–25 cm深段置于预先装满厌氧培养基的500 mL肖特瓶(厌氧培养瓶)中,并用丁基胶塞和顶空型试剂瓶盖密封。样品中有机碳含量和有机氮含量的测定委托中国有色桂林矿产地质研究院完成。
1.4 富集培养及产甲烷作用观测样品运回实验室后从原本装满培养基的肖特瓶中倒出110 mL上清液,并用氮气-二氧化碳混合气(4׃1,体积比)充满培养瓶顶部空间,最后再次用丁基胶塞和试剂瓶盖密封。
对于以乙酸盐为唯一能源物质的富集培养(富集物A),培养基中添加终浓度为20 mmol/L的乙酸钠;对于以氢气为唯一能源物质的富集培养(富集物H),培养瓶顶空间添加的是氮气-氢气-二氧化碳混合气(44׃11׃25,体积比));原位样品(富集物O)和富集培养组采用相同的培养基,但其中不含乙酸钠,顶空气中也不含氢气;阴性对照(富集物C)培养基中添加了终浓度为1 mol/L的NaOH,以杜绝其中的微生物活动。富集物A和富集物H包含3个重复,富集物C和富集物O分别只有一个样品。富集物A、富集物H和富集物C放置在28 ℃恒温避光培养,富集物O则在4 ℃避光保存,富集培养进行了21 d。
厌氧培养瓶顶部气体中甲烷的含量委托中国有色桂林矿产地质研究院通过气相色谱测定,待测气体使用气体采样器穿刺培养瓶上部丁基胶塞获得,气体样品被保存于气体采样袋。
1.5 DNA提取和基因文库构建富集培养后,所有样品的DNA分别被提取用作PCR的模板,富集物A和富集物H的模板DNA分别来自各自重复的混合。DNA提取使用土壤DNA提取试剂盒完成,操作步骤完全参照产品说明。
古菌16S rRNA基因序列通过PCR扩增获得,模板分别来自富集物A、富集物H和富集物O的DNA,引物为A8F (5′-CGGTTGATCCTGCCGGA-3′)和A1492R (5′-GGCTACCTTGTTACGACTT-3′)[24];PCR反应体系:模板DNA (10−20 ng/μL) 1−4 μL,上、下游引物(50 pmol/μL)各1 μL,dNTPs (2.5 mmol/L) 4 μL,牛血清白蛋白溶液(5 μg/μL) 1 μL,ExTaq DNA聚合酶(5 U/μL) 0.3 μL和10×ExTaq buffer 5 μL,加灭菌水使反应总体系至50 μL;PCR反应条件:95 ℃ 2 min;95 ℃ 30 s,51 ℃ 45 s,72 ℃ 60 s,37个循环;72 ℃ 7 min。
甲基辅酶M还原酶A亚基编码基因mcrA序列的PCR扩增采用引物ME1f (5′-GCMATGCARATH GGWATGTC-3′)和ME2r (5′-TCATKGCRTAGTTDG GRTAGT-3′)[25]。反应体系与前文相同。PCR反应条件:95 ℃ 2 min;95 ℃ 30 s,55 ℃ 45 s,72 ℃ 30 s,37个循环;72 ℃ 7 min。
PCR产物经过琼脂糖凝胶电泳检测后,目的片段通过胶回收试剂盒回收,胶回收片段被连接到pMD18-T载体上,连接产物被转化进入大肠杆菌DH5α感受态细胞,带有16S rRNA基因和mcrA基因片段的阳性克隆被分别挑选出来交由生工生物工程(上海)股份有限公司进行Sanger测序。
1.6 序列处理和文库多样性分析古菌16S rRNA基因序列长约1 490 bp,通过正反向测序结果拼接获得,序列拼接通过Geneious V10.2.6 (www.geneious.com)软件完成,共获得177条序列;mcrA基因全长约为1 650 bp,克隆文库中的序列为其中的一部分,长约750 bp,因此不需要拼接,共获得165条序列。
为了获得高质量的序列,所有序列都经过了嵌合体去除处理。嵌合体的分析采用UCHIME V4.2.4程序[26],古菌16S rRNA基因的参考序列来自Silva数据库(https://www.arb-silva.de)发布的核糖体RNA小亚基序列库,mcrA基因参考序列来自94个具有全基因组序列的产甲烷菌,数据来自GenBank (https://www.ncbi.nlm.nih.gov/genbank)。相似度≥95%的序列被归类为同一个操作分类单元(Operational taxonomic unit,OTU),使用Geneious V10.2.6完成。
文库覆盖率计算公式为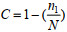
近源序列搜索采用BLASTp搜索GenBank完成(https://blast.ncbi.nlm.nih.gov/Blast.cgi),用于构建系统进化树的序列比对采用MAFFT V7.388[30],进化树采用FastTree V2.1.5[31]构建,所有序列的分析、处理和系统发育树的构建等操作均采用Geneious V10.2.6软件综合完成。
2 结果与分析 2.1 样品描述及产甲烷作用的观测用于富集培养的样品取自会仙湿地15 cm−23 cm深段沉积物,浅层样品呈灰色,深层样品呈黑色,有机碳量约为6.62% (干重,DW),孔隙水中氨氮量为6.98 mg/L,硝酸根离子含量小于0.072 mg/L,硫酸根含量为0.86−1.06 mg/L,钙离子含量为42−55 mg/L,pH约为7.47。有机质含量比较高、硝酸盐和硫酸盐含量比较低的环境被认为有利于产甲烷作用的发生。
富集培养过程中,分析了不同对照组培养瓶顶空气中甲烷的浓度(图 1):经过14 d培养,阴性对照(富集物C)和原位样品(富集物O)的顶空气中均未检测出甲烷(< 0.000 3 g/L),添加了氢气的富集培养(富集物H)和添加乙酸盐的富集培养(富集物A)在14 d时观测到甲烷浓度显著增加,富集物H和A中甲烷浓度平均值分别为0.06 g/m3和0.07 g/m3,两者无显著差异(P > 0.05)。以上结果表明,富集物H和富集物A中发生了微生物产甲烷活动。

|
| 图 1 不同富集培养物的顶空气甲烷含量 Figure 1 Headspace methane concentrations in different samples 注:H:氢型产甲烷富集条件的样品;A:乙酸型产甲烷富集条件的样品;O:未添加任何底物且置于4 ℃的原位样品;C:添加了氢氧化钠的阴性对照;ND:富集物O和富集物C顶空气中未检测到甲烷;仪器检测下限为0.000 3 g/m3,数据来源于3个重复的平均值. Note: H: H2-enriched sample; A: Acetate-enriched sample; O: Original sample without substrate supplementation and stored at 4 ℃; C: Negative control treated with 1 mol/L NaOH; ND: Methane was not detected in the headspace of either sample O or sample C; The detection limit was 0.000 3 g/m3; The presented data are averages from triplicate cultures. |
|
|
经过21 d富集培养,分别从富集物A、富集物H以及富集物O中提取了总DNA,针对古菌16S rRNA基因和甲基辅酶M还原酶A亚基编码基因mcrA基因,构建了6个基因文库,分别是:乙酸盐富集培养物的古菌16S rRNA基因文库AA,乙酸盐富集培养物的mcrA基因文库MA,氢气富集培养物的古菌16S rRNA基因文库AH,氢气富集培养物的mcrA基因文库MH,原位样品的古菌16S rRNA基因文库AO,以及原位样品的mcrA基因文库MO。经过PCR、克隆、测序结果分析和嵌合体排除等操作,最终从177个克隆子中获得146条高质量的古菌16S rRNA基因序列,从165个克隆子中得到138条高质量的mcrA基因序列,全部序列已经上传至GenBank,对应的登录号分别为MK075996−MK076122、MK123363−MK123380、MK123452和MK093263−MK093400。各文库中包含的序列总数、操作分类单元数以及多样性指数等参见表 1。
| 文库名称 Library name |
文库中克隆子数 Number of clones in the library |
操作分类单元数目 Number of OTUs |
辛普森指数 Simpson’s index (D) |
香农指数 Shannon’s index (H) |
覆盖率 Coverage (%) |
富集培养底物 Substrates for enrichment |
| AO-16S rRNA | 54 | 28 | 0.642 | 1.371 | 80 | 无 None |
| AH-16S rRNA | 41 | 19 | 0.629 | 1.314 | 78 | 氢气 Hydrogen |
| AA-16S rRNA | 51 | 22 | 0.380 | 0.818 | 84 | 乙酸盐 Acetate |
| MO-mcrA | 44 | 26 | 0.491 | 0.920 | 63 | 无 None |
| MH-mcrA | 46 | 19 | 0.132 | 0.292 | 80 | 氢气 Hydrogen |
| MA-mcrA | 48 | 20 | 0.120 | 0.234 | 85 | 乙酸盐 Acetate |
| 注:文库名字中首字母A代表古菌16S rRNA基因文库;首字母M代表mcrA基因文库. Note: The first letter “A” in the library name indicates a 16S rRNA gene library; The first letter “M” indicates an mcrA library. |
||||||
甲基辅酶M还原酶A亚基编码基因mcrA被认为是参与甲烷代谢相关古菌的特征基因,被广泛用于研究产甲烷古菌和甲烷厌氧氧化古菌的功能、数量和多样性[32]。从图 2可以看出,绝大部分mcrA序列可能都来自甲烷八叠球菌目,约占序列总数的85.5%,甲烷微球菌和ANME-1型厌氧甲烷氧化菌各占约5%,余下的4.3%来源于甲烷胞菌目的古菌,这与我们之前关于15 cm−25 cm深沉积物中mcrA基因来源组成的报道[22]基本一致。在图 2系统发育树中甲烷八叠球菌目又可细分为5个分支:Methanosarcina属一支;包含有ANME-2d型厌氧甲烷氧化菌(AD)的分支;包含有ZC-Ⅰ (Zoige clusterⅠ)类群古菌的分支;2个相对独立且不包含任何参考序列的分支,暂命名为KT-Ⅰ和KT-Ⅱ,在进化距离上,KT-Ⅰ相较于其他3个分支间的距离更远,KT-Ⅱ与AD较为接近。

|
| 图 2 McrA氨基酸序列的系统发育分析 Figure 2 Phylogenetic tree of McrA amino acid sequences 注:系统发育树由FastTree生成,参考序列的GenBank登录号列在参考物种名后括号中,OTU名称括号中的数字代表该OTU所包含的序列数,发育树中重要节点处的数值为可信度(Local support value,%),左下角标尺代表 20%的序列差异. Note: The phylogenetic tree was generated with FastTree program, the GenBank accessions of reference sequences are listed behind the names of species, the numbers within the parentheses of OTU tags indicate the number of sequences included in the OTU, the values on the important nodes represent the local support value (%), and the scale bars represent 20% sequence divergence. |
|
|
此次发现的138条mcrA序列中约有33%与我们之前报道的序列[22]一致(氨基酸序列相似度大于99.5%),余下的约67%是新发现的序列。新发现的序列中约有一半为KT-Ⅰ和KT-Ⅱ,这些序列相较于之前报道的序列而言,多样性更为丰富。在我们之前的报道[22]中,KT-Ⅰ只发现了1个序列,KT-Ⅱ为38条序列组成2个独立的OTU;而在本研究中,分支KT-Ⅰ由11个OTU (46条序列)组成,KT-Ⅱ包含了10个OTU (21条序列),详见图 2。
相较于原位样品MO而言,经过产甲烷条件富集培养后序列数量占比增幅最明显的是ZC-Ⅰ、AD和KT-Ⅱ三类古菌(图 3)。在文库MO中,这三类古菌的占比分别为11%、9%和11%;在氢型产甲烷条件下ZC-Ⅰ、AD和KT-Ⅱ类群古菌的序列占比分别为27%、16%和16%,增幅分别为145%、78%和45%;乙酸型产甲烷条件下ZC-Ⅰ、AD和KT-Ⅱ的序列占比分别为21%、23%和19%,增幅分别为90%、155%和72%。此外,KT-Ⅰ分支古菌的序列在MO、MH和MA三个mcrA基因文库中占比分别为35%、34%和31%,只有小幅改变(降幅3%–10%),而Methanosarcina属古菌仅出现在原位样品MO文库中。

|
| 图 3 产甲烷菌群落的物种组成 Figure 3 Taxonomic composition of methanogen communities 注:MO:原位样品;MH:氢气富集样品;MA:乙酸盐富集样品;KT-Ⅰ、KT-Ⅱ、ZC-Ⅰ和Methanosarcina属都是甲烷八叠球菌目的成员. Note: MO: Original sample; MH: H2 enrichment; MA: Acetate enrichment; KT-Ⅰ, KT-Ⅱ, ZC-Ⅰ and the Methanosarcina genus belong to the order Methanosarcinales. |
|
|
通过将文库中古菌16S rRNA基因序列在GenBank中进行比对,结果显示AO、AH和AA三个文库的全部146条序列中,145条序列与已报道古菌纯培养的序列相似度在81%−95%之间,只有一条序列(AO51)与Methanoregula formicica的相似度为97%。一般认为[33],同一个种之间的16S rRNA基因序列相似度应该≥97%,根据这一原则,本研究报道的16S rRNA基因序列中有145条来自古菌中尚未报道的新种。
为进一步认识文库中16S rRNA基因序列在古菌中的分类,用文库中69个OTU中的代表序列以及100条已报道的参考序列构建了系统发育树(图 4)。如图 4所示,16S rRNA基因文库中大部分的序列都分布在深古菌门(Bathyarchaeota)的树枝上(深古菌树枝包含了MCG-1和MCG-24等,共25个亚群),约占全部序列的92.4%,其余序列都分布在广古菌门(Euryarchaeota)的甲烷微菌目(Methanomicrobiales)和甲烷八叠球菌目(Methanosarcinales)所在的树枝上。

|
| 图 4 古菌16S rRNA基因的系统发育树 Figure 4 Phylogenetic tree of archaeal 16S rRNA gene sequences 注:系统发育树由FastTree生成;高亮显示的树枝包含了本研究发现的OTU;树枝的稳定性由可信度Local support value (%)表征,图中只在重要的内部节点处标注了Local support value (%);标尺代表 20%的序列差异;除MCG-11亚群外,建树用参照序列的名称及GenBank登录号在图中省略,MCG-20,MCG-21和MCG-22的参考序列出自之前的报道[42],其他参考序列的GenBank登录号为:AB087499,AB305017,NR_074531,NR_116054,KT453557,KT453558,NR_118849,AB454050,KP091065,AY592480,DQ301990,EF125500,AY396667,EF125508,EF680211,EU329775,DQ190101,DQ641821,EF125532,FJ901979,HM244168,EF367453,GQ848385,DQ302019,FJ264719,GQ848429,FJ175640,GU475241,AB362547,EU385859,AY354121,FJ902705,FJ264803,FJ404037,FJ649521,EU329812,EU329816,EU329843,AB177007,DQ301970,AB801039,DQ301992,DQ301965,AB213086,AB213075,AB213083,AY555815,EU924228,FJ936660,KP784731,AB113631,AB113635,EF367492,FJ649517,FJ649514,FJ902285,AB300143,FJ853472,FJ901443,AB161328,FJ716366,FJ821625,GU257243,HM244089,AB094546,AB161338,AB426445,AM712527,AY396665,AY454600,AY464796,AY591992,AY861924,EU635904,HM483669,AB015273,EF367607,FJ901444,HM244179,NR_134838,NR_074192,AB301476,AJ458436,JN971012,KU290365,JN244020,AY351467,FJ982666,AB479411,AB669269,GU135467,JX266091,JX648297,HQ700677,DQ084452,HM041907. Note: The phylogenetic tree was generated with FastTree program; Branches containing sequences identified in this study are highlighted in different colors; The stability of the tree is evaluated by using the local support value (%), and local support values are shown only for important nodes; The scale bars represent 20% sequence divergence; The names and GenBank accession of the reference sequences are not shown except for MCG-11; Reference sequences of MCG-20, MCG-21 and MCG-22 are from the previous report [42], and for the rest of the reference sequences, their GenBank accessions are listed below: AB087499, AB305017, NR_074531, NR_116054, KT453557, KT453558, NR_118849, AB454050, KP091065, AY592480, DQ301990, EF125500, AY396667, EF125508, EF680211, EU329775, DQ190101, DQ641821, EF125532, FJ901979, HM244168, EF367453, GQ848385, DQ302019, FJ264719, GQ848429, FJ175640, GU475241, AB362547, EU385859, AY354121, FJ902705, FJ264803, FJ404037, FJ649521, EU329812, EU329816, EU329843, AB177007, DQ301970, AB801039, DQ301992, DQ301965, AB213086, AB213075, AB213083, AY555815, EU924228, FJ936660, KP784731, AB113631, AB113635, EF367492, FJ649517, FJ649514, FJ902285, AB300143, FJ853472, FJ901443, AB161328, FJ716366, FJ821625, GU257243, HM244089, AB094546, AB161338, AB426445, AM712527, AY396665, AY454600, AY464796, AY591992, AY861924, EU635904, HM483669, AB015273, EF367607, FJ901444, HM244179, NR_134838, NR_074192, AB301476, AJ458436, JN971012, KU290365, JN244020, AY351467, FJ982666, AB479411, AB669269, GU135467, JX266091, JX648297, HQ700677, DQ084452, HM041907. |
|
|
16S rRNA基因文库中来自广古菌门产甲烷菌的序列共有11条,其中9条序列属于甲烷八叠球菌目,2条来自甲烷微菌目。甲烷八叠球菌的序列数量在文库AO和AH中分别占7%和12%,甲烷微菌目序列仅在文库AO中出现,约占4%,文库AA中没有检测到产甲烷菌的16S rRNA基因序列。这些序列(AO51除外)与已报道的16S rRNA基因序列相似度在87%−95%之间,可能来自已产甲烷菌的新种。
3 讨论 3.1 岩溶环境氢型产甲烷菌和乙酸型产甲烷菌的主要类群由富集培养过程中甲烷的检出以及基因文库中古菌群落组成的变化看来,主要的产甲烷菌类群应该是甲烷八叠球菌目的古菌。
值得注意的是,AA文库(乙酸型产甲烷条件的古菌16S rRNA基因文库)中没有检测到甲烷八叠球菌。这可能是由下列4个原因造成的:(1)古菌通用引物A8F和A1492R与深古菌的结合效果更好,产生扩增偏好性;(2) AA文库的覆盖率为84%,尚有一部分古菌未被覆盖;(3)培养时间不足,产甲烷菌数量没有得到显著的提升;(4) MCG-11等深古菌的数量增加了。基于以上原因,产甲烷菌在群落中的占比下降,因此没有检测到甲烷八叠球菌。
3.2 甲烷八叠球菌目古菌的主要类型及代谢潜能已知的甲烷八叠球菌目包含氢型、乙酸型、混合型产甲烷菌以及厌氧甲烷氧化古菌(Anaerobic methanotrophic archaea,ANME),除了能利用乙酸、氢气和二氧化碳产甲烷外,还可以利用甲醇、甲胺和甲烷等多种类型底物[34]。本研究中甲烷八叠球菌目的古菌是3个mcrA文库中最主要的古菌类群,主要包括ZC-Ⅰ、AD、KT-Ⅰ和KT-Ⅱ四个分支,在2种产甲烷条件下序列占比都有显著提升的是ZC-Ⅰ、AD和KT-Ⅱ三个类群。
ZC-Ⅰ分支最早被发现于西藏若尔盖(Zoige)湿地,在我国洪泽湖、鄱阳湖和三江平原等多地也有分布[35],被认为是能够代谢乙酸盐和氢气的产甲烷菌类群[36];本研究中经过富集培养后,ZC-Ⅰ序列在文库MH和MA中的占比分别增加了145%和90%,表明岩溶环境的ZC-Ⅰ可能也具备乙酸型和氢型产甲烷能力。
AD类群McrA氨基酸序列在系统进化树(图 2)中与厌氧甲烷氧化古菌中的ANME-2d类群位于同一个分支,ANME中的许多成员都具有mcrA基因,被认为能通过逆向的产甲烷途径进行甲烷厌氧氧化,也有研究表明部分ANME可能同时具备甲烷产生和甲烷氧化的能力[37-38]。AD类群在氢型和乙酸型产甲烷条件下都有较为显著的增加,表明其具有潜在的产甲烷能力,但由于ANME-2d古菌也可利用沉积物中腐殖酸等物质进行甲烷氧化[39],因此AD分支古菌在产甲烷条件下的具有代谢活动有待深入研究。
KT-Ⅰ和KT-Ⅱ是系统发育树上相对独立的分支(图 2),截至本文撰写时(2018年12月),暂未发现任何已报道的序列能够分布在这2个分支中,其代谢方式也没有任何资料可供参考,经过富集培养后KT-Ⅰ在2种富集物中的组成变化幅度较小,而KT-Ⅱ在经过氢型产甲烷条件和乙酸型产甲烷条件培养后,其序列占比由原位样品中的11%分别增加至16%和19%,暗示着KT-Ⅱ类群古菌可能同时包含能够利用氢气和乙酸的产甲烷菌类型。
3.3 产甲烷条件下深古菌的群落组成、代谢方式及与产甲烷菌的相互关系深古菌最初被命名为陆地型MCG古菌(Terrestrial miscellaneous crenarchaeotal group)[40],于2014年被正式命名为Bathyarchaeota (深古菌)[41],是本研究16S rRNA基因文库中最丰富的古菌类群。根据最新的分类方法[42],深古菌门内又被进一步划分为25个亚群,如图 4所示MCG-1–MCG-24,其中MCG-5分为MCG-5a和MCG-5b两个亚群。本文所发现的深古菌序列主要分布在MCG-11 (约占总序列的73.3%)、MCG-6 (9.6%)、MCG-5b (4.1%)以及MCG-9 (0.7%)的树枝上,还有一部分序列命名为“Unclassified MCG” (4.8%)尚无法确定其在深古菌门中的分类(图 4)。
目前为止尚无深古菌可以在实验室进行纯培养的报道,基于非培养研究方法获得的结果揭示了部分深古菌的代谢潜力[42-43]:例如,乙酸盐被证明可以被MCG-8和MCG-15亚群深古菌用作生物合成DNA等物质[44-45],氢气可被深古菌中的一些类群(MCG-13和MCG-21)用于二氧化碳固定和产乙酸[42, 46],一部分深古菌(来自MCG-3和MCG-8)被认为可能具有厌氧甲烷氧化的能力[8]。
本研究中没有发现MCG-8等深古菌,但观察到MCG-11和Unclassified MCG的序列占比在乙酸盐富集培养条件下显著增加,MCG-6、MCG-9和Unclassified MCG三类深古菌的序列在氢型产甲烷条件下有较为显著的增加(图 5),显示出它们对乙酸盐和氢气潜在的代谢能力。另外,由于各mcrA文库中都发现了甲烷以及数量可观的ANME-1和ANME-2d型厌氧甲烷氧化菌的mcrA基因序列,它们有可能利用了沉积物中的有机质作为电受体进行厌氧甲烷氧化[39],因此也不能排除培养物中的部分深古菌采用类似的厌氧甲烷氧化途径获得能量的可能性。

|
| 图 5 古菌群落的物种组成 Figure 5 Taxonomic composition of archaeal communities 注:AO代表原位样品的古菌;AH代表氢气富集样品;AA代表乙酸盐富集样品;MCG-5b、–6、–9和–11都是深古菌门中的亚群. Note: AO represents the original sample; AH is the H2-enriched enrichment; AA stands for the acetate-enriched enrichment; The MCG-5b, –6, –9 and –11 are the subgroups belonging to the Bathyarchaeota phylum. |
|
|
会仙岩溶湿地是一个有机质含量比较高、硝酸盐和硫酸盐含量比较低的岩溶环境,湿地沉积物中的古菌以深古菌和广古菌门的产甲烷菌为主,其中蕴涵着许多新型古菌。
实验室培养条件下会仙岩溶湿地沉积物样品中主要的氢型产甲烷菌和乙酸型产甲烷菌都来自甲烷八叠球菌目,主要包括Zoige ClusterⅠ (ZC-Ⅰ)、ANME-2d Cluster (AD)、以及新类群KT-Ⅰ和KT-Ⅱ四个分支。
以MCG-11亚群为主的深古菌是会仙岩溶湿地沉积物样品中占比最高的古菌类群,在产甲烷培养条件下仍然能在古菌群落中维持优势,表明深古菌具有多样的代谢潜能和环境适应性,可能在岩溶湿地环境及产甲烷条件下都发挥着重要的作用。
| [1] |
Palmer AN. Origin and morphology of limestone caves[J]. GSA Bulletin, 1991, 103(1): 1-21. DOI:10.1130/0016-7606(1991)103<0001:OAMOLC>2.3.CO;2 |
| [2] |
Hartmann A, Goldscheider N, Wagener T, et al. Karst water resources in a changing world: Review of hydrological modeling approaches[J]. Reviews of Geophysics, 2014, 52(3): 218-242. DOI:10.1002/2013RG000443 |
| [3] |
Reitner J, Thiel V. Encyclopedia of Geobiology[M]. Dordrecht: Springer, 2011: 521.
|
| [4] |
White WB, Culver DC. Encyclopedia of Caves[M]. Waltham: Academic Press, 2012: 81.
|
| [5] |
Conrad R. The global methane cycle: recent advances in understanding the microbial processes involved[J]. Environmental Microbiology Reports, 2009, 1(5): 285-292. DOI:10.1111/j.1758-2229.2009.00038.x |
| [6] |
Conrad R. Contribution of hydrogen to methane production and control of hydrogen concentrations in methanogenic soils and sediments[J]. FEMS Microbiology Ecology, 1999, 28(3): 193-202. DOI:10.1111/j.1574-6941.1999.tb00575.x |
| [7] |
Conrad R, Claus P. Contribution of methanol to the production of methane and its 13C-isotopic signature in anoxic rice field soil[J]. Biogeochemistry, 2005, 73(2): 381-393. DOI:10.1007/s10533-004-0366-9 |
| [8] |
Evans PN, Parks DH, Chadwick GL, et al. Methane metabolism in the archaeal phylum Bathyarchaeota revealed by genome-centric metagenomics[J]. Science, 2015, 350(6259): 434-438. DOI:10.1126/science.aac7745 |
| [9] |
Vanwonterghem I, Evans PN, Parks DH, et al. Methylotrophic methanogenesis discovered in the archaeal phylum Verstraetearchaeota[J]. Nature Microbiology, 2016, 1(12): 16170. DOI:10.1038/nmicrobiol.2016.170 |
| [10] |
Schirmack J, Mangelsdorf K, Ganzert L, et al. Methanobacterium movilense sp. nov., a hydrogenotrophic, secondary-alcohol-utilizing methanogen from the anoxic sediment of a subsurface lake[J]. International Journal of Systematic and Evolutionary Microbiology, 2014, 64(2): 522-527. |
| [11] |
Ganzert L, Schirmack J, Alawi M, et al. Methanosarcina spelaei sp. nov., a methanogenic archaeon isolated from a floating biofilm of a subsurface sulphurous lake[J]. International Journal of Systematic and Evolutionary Microbiology, 2014, 64(10): 3478-3484. |
| [12] |
McDonough LK, Iverach CP, Beckmann S, et al. Spatial variability of cave-air carbon dioxide and methane concentrations and isotopic compositions in a semi-arid karst environment[J]. Environmental Earth Sciences, 2016, 75(8): 700. DOI:10.1007/s12665-016-5497-5 |
| [13] |
Fillol M, Sànchez-Melsió A, Gich F, et al. Diversity of Miscellaneous crenarchaeotic group archaea in freshwater karstic lakes and their segregation between planktonic and sediment habitats[J]. FEMS Microbiology Ecology, 2015, 91(4): fiv020. |
| [14] |
Kučinskienė A, Krevš A. Mineralization of organic matter in bottom sediments of the littoral zones of four Lithuanian lakes[J]. Ekologija, 2006(1): 40-47. |
| [15] |
Chen Y, Wu LQ, Boden R, et al. Life without light: microbial diversity and evidence of sulfur- and ammonium-based chemolithotrophy in Movile Cave[J]. The ISME Journal, 2009, 3(9): 1093-1104. DOI:10.1038/ismej.2009.57 |
| [16] |
Tetu SG, Breakwell K, Elbourne LDH, et al. Life in the dark: metagenomic evidence that a microbial slime community is driven by inorganic nitrogen metabolism[J]. The ISME Journal, 2013, 7(6): 1227-1236. DOI:10.1038/ismej.2013.14 |
| [17] |
Ortiz M, Legatzki A, Neilson JW, et al. Making a living while starving in the dark: metagenomic insights into the energy dynamics of a carbonate cave[J]. The ISME Journal, 2014, 8(2): 478-491. DOI:10.1038/ismej.2013.159 |
| [18] |
Reitschuler C, Sp tl C, Hofmann K, et al. Archaeal distribution in moonmilk deposits from alpine caves and their ecophysiological potential[J]. Microbial Ecology, 2016, 71(3): 686-699. DOI:10.1007/s00248-015-0727-z |
| [19] |
Legatzki A, Ortiz M, Neilson JW, et al. Bacterial and archaeal community structure of two adjacent calcite speleothems in Kartchner Caverns, Arizona, USA[J]. Geomicrobiology Journal, 2011, 28(2): 99-117. DOI:10.1080/01490451003738465 |
| [20] |
Borrel G, Jézéquel D, Biderre-Petit C, et al. Production and consumption of methane in freshwater lake ecosystems[J]. Research in Microbiology, 2011, 162(9): 832-847. DOI:10.1016/j.resmic.2011.06.004 |
| [21] |
Ma ZL, Cai DS, Jiang ZC. About Karst wetland classification system[J]. Journal of Guangxi Normal University (Natural Science Edition), 2009, 27(2): 101-106. (in Chinese) 马祖陆, 蔡德所, 蒋忠诚. 岩溶湿地分类系统研究[J]. 广西师范大学学报:自然科学版, 2009, 27(2): 101-106. |
| [22] |
Chen Y, Qiu KR, Wu Q, et al. Methanogenic community structure, abundance, and activity in Huixian karst wetland, Guilin, China[J]. Chinese Journal of Applied & Environmental Biology, 2017, 23(6): 959-967. (in Chinese) 陈颖, 邱凯瑞, 吴麒, 等. 桂林会仙岩溶湿地产甲烷菌的数量、群落组成和活性[J]. 应用与环境生物学报, 2017, 23(6): 959-967. |
| [23] |
Wolfe RS. Techniques for cultivating methanogens[J]. Methods in Enzymology, 2011, 494: 1-22. DOI:10.1016/B978-0-12-385112-3.00001-9 |
| [24] |
Casamayor EO, Sch fer H, Ba eras L, et al. Identification of and spatio-temporal differences between microbial assemblages from two neighboring sulfurous lakes: comparison by microscopy and denaturing gradient gel electrophoresis[J]. Applied and Environmental Microbiology, 2000, 66(2): 499-508. DOI:10.1128/AEM.66.2.499-508.2000 |
| [25] |
Hales BA, Edwards C, Ritchie DA, et al. Isolation and identification of methanogen-specific DNA from blanket bog peat by PCR amplification and sequence analysis[J]. Applied and Environmental Microbiology, 1996, 62(2): 668-675. |
| [26] |
Edgar RC, Haas BJ, Clemente JC, et al. UCHIME improves sensitivity and speed of chimera detection[J]. Bioinformatics, 2011, 27(16): 2194-2200. DOI:10.1093/bioinformatics/btr381 |
| [27] |
Mullins TD, Britschgi TB, Krest RL, et al. Genetic comparisons reveal the same unknown bacterial lineages in Atlantic and Pacific bacterioplankton communities[J]. Limnology and Oceanography, 1995, 40(1): 148-158. DOI:10.4319/lo.1995.40.1.0148 |
| [28] |
Simpson EH. Measurement of diversity[J]. Nature, 1949, 163(4148): 688. DOI:10.1038/163688a0 |
| [29] |
Shannon CE. A mathematical theory of communication[J]. ACM SIGMOBILE Mobile Computing and Communications Review, 2001, 5(1): 3-55. DOI:10.1145/584091.584093 |
| [30] |
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability[J]. Molecular Biology and Evolution, 2013, 30(4): 772-780. DOI:10.1093/molbev/mst010 |
| [31] |
Price MN, Dehal PS, Arkin AP. FastTree 2 – Approximately maximum-likelihood trees for large alignments[J]. PLoS One, 2010, 5(3): e9490. DOI:10.1371/journal.pone.0009490 |
| [32] |
Grabarse W, Shima S, Mahlert F, et al. Methyl-coenzyme M reductase[J]. New York: Wiley, 2011, 19. |
| [33] |
Yarza P, Yilmaz P, Pruesse E, et al. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences[J]. Nature Reviews Microbiology, 2014, 12(9): 635-645. DOI:10.1038/nrmicro3330 |
| [34] |
Zhang JC, Xu YQ, Lu YH. Microbial mechanisms of methane production and oxidation in terrestrial ecosystems[J]. Acta Ecologica Sinica, 2015, 35(20): 6592-6603. (in Chinese) 张坚超, 徐镱钦, 陆雅海. 陆地生态系统甲烷产生和氧化过程的微生物机理[J]. 生态学报, 2015, 35(20): 6592-6603. |
| [35] |
Liu DY, Ding WX, Jia ZJ, et al. The impact of dissolved organic carbon on the spatial variability of methanogenic archaea communities in natural wetland ecosystems across China[J]. Applied Microbiology and Biotechnology, 2012, 96(1): 253-263. DOI:10.1007/s00253-011-3842-x |
| [36] |
Zhang GS, Tian JQ, Jiang N, et al. Methanogen community in Zoige wetland of Tibetan plateau and phenotypic characterization of a dominant uncultured methanogen cluster ZC-Ⅰ[J]. Environmental Microbiology, 2008, 10(7): 1850-1860. DOI:10.1111/j.1462-2920.2008.01606.x |
| [37] |
Bertram S, Blumenberg M, Michaelis W, et al. Methanogenic capabilities of ANME-archaea deduced from 13C-labelling approaches[J]. Environmental Microbiology, 2013, 15(8): 2384-2393. DOI:10.1111/1462-2920.12112 |
| [38] |
Wegener G, Krukenberg V, Ruff SE, et al. Metabolic capabilities of microorganisms involved in and associated with the anaerobic oxidation of methane[J]. Frontiers in Microbiology, 2016, 7: 46. |
| [39] |
Valenzuela EI, Prieto-Davó A, López-Lozano NE, et al. Anaerobic methane oxidation driven by microbial reduction of natural organic matter in a tropical wetland[J]. Applied and Environmental Microbiology, 2017, 83(11): e00645-17. |
| [40] |
Takai S, Henton MM, Picard JA, et al. Prevalence of virulent Rhodococcus equi in isolates from soil collected from two horse farms in South Africa and restriction fragment length polymorphisms of virulence plasmids in the isolates from infected foals, a dog and a monkey[J]. Onderstepoort Journal of Veterinary Research, 2001, 68(2): 105-110. |
| [41] |
Meng J, Xu J, Qin D, et al. Genetic and functional properties of uncultivated MCG archaea assessed by metagenome and gene expression analyses[J]. The ISME Journal, 2014, 8(3): 650-659. DOI:10.1038/ismej.2013.174 |
| [42] |
Zhou ZC, Pan J, Wang FP, et al. Bathyarchaeota: globally distributed metabolic generalists in anoxic environments[J]. FEMS Microbiology Reviews, 2018, 42(5): 639-655. DOI:10.1093/femsre/fuy023 |
| [43] |
Chen YL, Pan J, Zhou ZC, et al. Progress in studies on Bathyarchaeota in coastal ecosystems[J]. Microbiology China, 2017, 44(7): 1690-1698. (in Chinese) 陈玉连, 潘杰, 周之超, 等. 滨海深古菌的研究进展[J]. 微生物学通报, 2017, 44(7): 1690-1698. |
| [44] |
Webster G, Rinna J, Roussel EG, et al. Prokaryotic functional diversity in different biogeochemical depth zones in tidal sediments of the Severn Estuary, UK, revealed by stable-isotope probing[J]. FEMS Microbiology Ecology, 2010, 72(2): 179-197. |
| [45] |
Na H, Lever MA, Kjeldsen KU, et al. Uncultured Desulfobacteraceae and crenarchaeotal group C3 incorporate 13C-acetate in coastal marine sediment[J]. Environmental Microbiology Reports, 2015, 7(4): 614-622. DOI:10.1111/1758-2229.12296 |
| [46] |
He Y, Li M, Perumal V, et al. Genomic and enzymatic evidence for acetogenesis among multiple lineages of the archaeal phylum Bathyarchaeota widespread in marine sediments[J]. Nature Microbiology, 2016, 1(6): 16035. DOI:10.1038/nmicrobiol.2016.35 |