 2019, Vol. 46
2019, Vol. 46扩展功能
文章信息
- 李还原, 董志祥, 陈奕霏, 郭军
- LI Huan-Yuan, DONG Zhi-Xiang, CHEN Yi-Fei, GUO Jun
- 蜜蜂肠道菌群定殖研究进展
- Advances in research of colonization of gut microbiota in the honeybee
- 微生物学通报, 2019, 46(11): 3091-3101
- Microbiology China, 2019, 46(11): 3091-3101
- DOI: 10.13344/j.microbiol.china.190015
-
文章历史
- 收稿日期: 2019-01-03
- 接受日期: 2019-05-05
- 网络首发日期: 2019-05-29





动物从进化起源开始就接触微生物,从而产生亲密的微生物-宿主相互作用,不同的微生物群落定殖于不同的宿主组织中,而胃肠道是肠道菌群分布最密集和多样化最高的区域[1]。蜜蜂与其他社会性动物都是通过相互接触来获取和传播肠道细菌,并在长期的遗传和进化过程中形成了特异的、功能专一的菌群。这种社会性接触传播可能建立稳定的、长期的微生物群,并与宿主产生相互适应的共生关系[2]。
蜜蜂(Apis mellifera)作为全球最重要的传粉昆虫,和其他野生传粉蜂类(如熊蜂)在全球生态系统服务和人类农业中发挥了关键作用[3]。蜜蜂肠道是消化、食物加工、营养吸收和供给的主要部位,也是各种病原菌如微孢子虫(Nosema ceranae)[4]及大部分的蜜蜂病毒[5]的感染位点。蜜蜂肠道中的有益菌群对各种压力源的调节作用在一定程度上提高了蜂群的抵抗力,如肠道益生菌能间接提高寄主的免疫能力,或者通过种间竞争抑制病原微生物的发展[6]。了解和研究健康蜂群的肠道细菌的定殖和演替规律,可为蜜蜂养殖的肠道生理健康标准提供理论依据,对深入了解蜜蜂肠道微生态系统平衡调控具有重要意义。本文综述了蜜蜂肠道菌群的特征及菌群定殖研究进展,对影响蜜蜂肠道菌群定殖的影响因素进行了介绍,并对宿主的功能和新陈代谢对肠道菌群的影响进行了探讨。
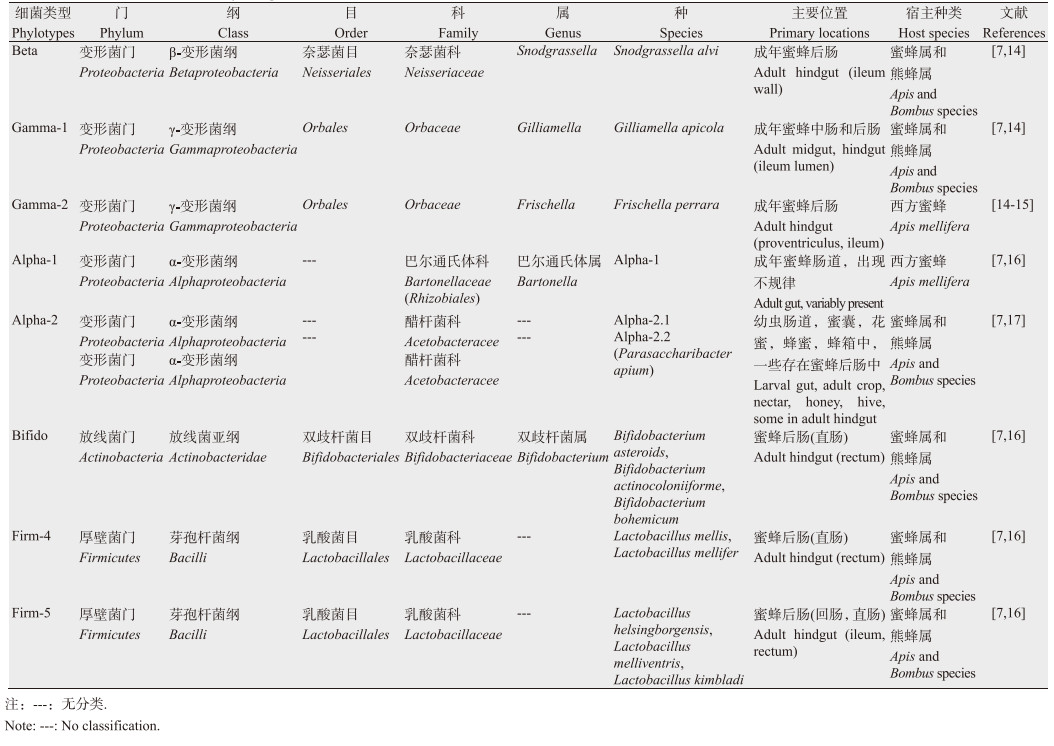
1 蜜蜂肠道菌群的特征 1.1 蜂群肠道核心菌群的组成及分类地位蜜蜂肠道菌群具有高度保守性,基于高通量测序技术对西方蜜蜂肠道菌群多样性的研究揭示出蜜蜂肠道菌群主要由8大类9种细菌组成[7-11]。这8大类细菌主要包括β-变形菌纲的Snodgrassella alvi,γ-变形菌纲的Gilliamella apicola和Frischella perrara,放线菌门的Bifidobacterium asteroides,厚壁菌门的Firm-4、Firm-5,以及α-变形菌纲的Alpha-1、Alpha-2.1和Alpha-2.2[7, 12-13] (表 1),这些细菌已经在全球不同地区的西方蜜蜂(A. mellifera)肠道中被发现[7-10, 18-19]。在东方蜜蜂(A. cerana)[11, 20-21]和熊蜂属(Bombus)的很多种中[11, 22]也发现了这些细菌类群及亲缘关系密切的菌属。蜜蜂主要通过社会传播获得这些细菌,其中S. alvi、G. apicola、Firm-4、Firm-5、B. asteroides这5种在蜜蜂肠道中比较常见,在熊蜂肠道中也存在这5种细菌[23],这5种细菌构成了蜜蜂的核心肠道菌群(95%以上)。
蜜蜂肠道菌群归属于9种高度特异的细菌,并且长期保持稳定,具有社会性的熊蜂和其他蜂种也具有这些细菌;然而,迄今为止,在独居蜂和无粉框的蜂类中尚未发现含有这种特殊的微生物群落[18-20]。蜜蜂特有的一种拟杆菌属Apibacter adventoris在蜜蜂属和熊蜂属中均被发现,与蜜蜂肠道核心菌群相比,它在西方蜜蜂中的丰度一直很低[9, 21-24],系统进化分析表明它们属于一类特定蜜蜂肠道细菌。其他细菌谱系似乎受到宿主的限制,如Bartonella apis和F. perrara只在Apis spp.中找到;肠道共生菌Schmidhempelia bombi[25]与G. apicola相关;Bombiscardovia cosgulans[26]主要在熊蜂属中被发现;另外,在蜜蜂属和熊蜂属中发现了不同的双歧杆菌属[27]。
不同的宿主物种可以携带不同的细菌菌株,相关的宿主往往会携带相关的菌株;这意味着在漫长的进化时间尺度上存在着多样化和共同适应性[28]。这种特异性可能因不同的共生菌不能跨越宿主定殖的屏障作用而得到加强。例如,S. alvi菌株的移植实验显示,从蜜蜂中分离出的菌株不能在熊蜂中定殖,反之亦然[29-30]。然而,特殊细菌与通用性细菌(那些能够在蜜蜂物种以及更广阔的环境中定居的细菌)对肠道整体微生物群落结构和功能的贡献尚不清楚。L. kunkeei的比较基因组学研究发现,L. kunkeei在花蜜和蜂房材料中很常见,但在肠道中很少见,表明它经常在宿主物种之间移动,并且没有显示出共生多样性的证据[31]。
在蜜蜂肠道以外的环境中一直没有发现专属的核心肠道细菌。这种限制可能是强制的,部分原因是由于这些细菌在大气中的高氧浓度中不能繁殖。核心肠道细菌物种主要是兼性厌氧菌或微型嗜热菌组成,因此在很大程度上取决于宿主间的相互作用。这种相互作用类似于哺乳动物肠道微生物群传播的途径,反映在蜜蜂的整个生命期内肠道微生物群的发展模式。
1.3 蜜蜂肠道菌群的定殖规律及影响因素肠道菌群的定殖是一个渐进的过程,蜜蜂出房时肠道菌群很少[10],随着日龄的增长,通过交哺、采食等方式与外界的细菌接触机会增加,为各种细菌在肠道内定殖提供了有利条件,并逐步形成稳定的微生态平衡系统。而当肠道中正常的微生物群落受到宿主及外界环境的影响后,有害细菌可能趁虚而入,打破原有的微生物之间的动态平衡关系,从而导致肠道菌群失调而引起宿主患病。
1.3.1 蜜蜂肠道菌群定殖与年龄、群体和季节变化的关系(1) 蜜蜂日龄对肠道菌群定殖的影响。在幼虫到蛹和成年蜜蜂的变态发育过程中,由于肠内脏会出现脱落,新出房的成年蜜蜂很少或没有肠道细菌,随着新出房工蜂与箱内环境的接触及工蜂的哺育,肠道菌群开始正常的定殖[12, 29]。基于qPCR (Real-time quantitative PCR detecting system)方法对西方蜜蜂肠道菌群的定殖过程的研究表明,幼虫和刚出房的工蜂几乎不包含细菌,从4-6日龄开始,蜜蜂肠道中特定细菌群落开始大量增加,且6日龄以后西方蜜蜂中肠道菌群保持稳定[10]。而对本土中华蜜蜂(A. c. cerana,简称中蜂)的研究表明,1日龄的中蜂肠道中细菌很少,其中乳酸菌的数量相对较多,Gilliamella、Bifidobacterium和Snodgrassella的操作分类单元(Operational taxonomic units,OTU)出现的次数分别为174、85和35次(占OTU总数的百分比分别为0.75%、0.37%和0.15%),由于其含量较低,因此qPCR方法较难检测到[32],这也证实成年蜂中常见的共生菌在1日龄幼蜂中的确存在,蜜蜂体内的优势菌群是从1日龄开始不断积累增加的过程。新出房工蜂(Newly emerged bees,NEB)前3日龄的主要任务就是反复清理巢房等简单的箱内活动[33],在出房3日后通常出现一次行为的转变,即从清理巢房转变到哺育幼蜂的工作中[33-34];工蜂3日龄时,微生物群落中含有大于107个细菌,这些细菌大多数属于蜜蜂肠道的特征种类,并且回肠和直肠开始显示“正常”的群落组成。Anderson等[34]的报道表明西方蜜蜂第7日龄工蜂的细菌类型与成年工蜂几近一致。第8日,这些微生物群落稳定在大约109个细菌。肠道菌群一旦建立起来,当工蜂在不同的行为阶段过渡时,肠道微生物群落基本稳定[35]。而对中蜂(A. c. cerana)研究发现,肠道菌群受宿主日龄的影响并处于动态变化之中[36],且在我国兰州熊蜂的研究中也存在相似的变化趋势[11]。另外,Ahn等[21]对韩国的东方蜜蜂(A. c. indica)不同发育阶段的肠道菌群的多样性研究也证实了蜜蜂肠道菌群也是有少数几个特定细菌种群占主要地位。
蜜蜂肠道菌群在出巢采集前已经稳定定殖,这意味着工蜂可在蜂箱环境中通过巢房或巢房中的组分(如蜂蜡)传播细菌。实验也证实,当蜜蜂直接接触到年长的蜜蜂或被喂食用后肠浸渍过的糖水时,最典型的肠道微生物群落定殖发生的可能性最高[12]。研究还发现,蜜蜂的交哺行为不是肠道细菌传播的主要途径,这与前肠含有少量细菌的研究结果相一致[10, 13]。尽管蜜蜂可通过接触活蜂获得常见的微生物,但蜜蜂还能通过粪口途径传播肠道细菌,特别是S. alvi、G. apicola和F. perrara等细菌可能通过这一途径来获取[12, 34],而蜂群中贮存的花粉(蜂粮)在传播醋杆菌科的细菌中发挥了重要的作用[37]。
(2) 蜜蜂群体对肠道菌群定殖的影响。通过测序分析得知,成年蜂王与工蜂的肠道微生物群落存在显着差异,蜂王肠道微生物群落的大小和组成与工蜂各不相同。蜂王肠道微生物群落在生命早期以肠道细菌为主,但成熟期主要由α-变形杆菌(Alphaproteobacteria)组成;此外,蜂王肠道菌群并未表现出工蜂肠道菌群稳定的趋势[38],往往缺乏某些特殊种类的细菌。肠道菌群组成的差异可能是由于蜂王独特的生理和饮食结构所致,如蜂王主要以工蜂分泌的高营养的蜂王浆为主。工蜂的肠道菌群,主要由3个门的细菌组成(Firmicutes、Proteobacteria和Actinobacteria),它们在蜜蜂的肠道中具有转录活性[39]。成年工蜂的核心微生物群的特征是由少量的细菌分支组成,一些属于新命名的新属或种[7, 14-15]。这些核心分支被称为Firm-4、Firm-5、Bifido、Alpha-2.1、Alpha-2.2、Alpha-1、Beta、Gamma-1和Gamma-2[7]。熊蜂中Snodgrassella属(S. alvi)细菌含量最高,而S. alvi已被证实在熊蜂上具有提高宿主蜂对寄生的短膜虫(Crithidia bombi)的抵抗作用[6]。
(3) 季节变化对肠道菌群定殖的影响。国外一些研究者也对蜜蜂肠道菌群的季节性变化做了一定的探讨,Hroncova等[40]分析了西方蜜蜂的肠道菌群后发现在特定的时间点,这些肠道菌群可能受到季节性节律的影响,不同年份的观察结果也发现具有较高的变异;在秋季和春季期间,蜜蜂之间的整个肠道内的微生物群落会出现微小差异。偶尔,在成年工蜂个体中,肠道微生物群落的正常组成也可能存在很大偏差。关于季节性趋势,对工蜂中肠的研究发现,微生物群落在6个月内发生了变化[41]。另一项研究在油菜花期通过对3个蜂种A. m. carnica、Osmia bicornis和B. terrestris进行连续3年的观察发现,它们的肠道菌群在不同年份间差异显著[42]。Ludvigsen[43]的研究表明,共生菌G. apicola的数量随着季节的变化而变化,其出现的频数在一年内持续的下降,并在十月份的蜜蜂样品中数量最低,而S. alvi在这个月份数量开始上升。在一些年份,肠道菌群在单群蜂和不同群蜜蜂个体上具有较大的变异。而东方蜜蜂的相关研究则发现这两种细菌在不同季节间保持稳定,差异并不显著[32]。最近的一项研究也发现采集蜂肠道内共生菌Firm-5从春季到秋季大约下降了25%[13],但我们对东方蜜蜂肠道内乳酸菌的检测发现,乳酸菌的含量从春季到秋季处于一个上升的趋势[32]。这种研究结果的差异,很可能是所采用的测序方法的不同引起的,也可能是受蜂种、地理区域等多因素综合影响所致。
1.3.2 蜜蜂不同肠道部位菌群定殖规律昆虫为适应各种特殊生态位和取食习惯,经过长期协同进化逐渐演化为特定肠道部位定居特定肠道微生物的现象[44]。蜜囊是肠道内的一种肌肉质的可膨胀器官,以适应采集蜂采集花蜜[45-46]。虽然蜜囊含有营养丰富的花蜜并可作为微生物的能量来源,qPCR及FISH (Fluorescence in situ hybridization)染色结果表明蜜囊内几乎不包含有细菌[10],但其他学者从蜜囊中分离出13种乳酸菌[47]和部分双歧杆菌[48]菌株,这些细菌也可能是来自花蜜而非蜜蜂体内共生的菌群。蜜囊这种频繁的填充和排空采集的花蜜到蜂箱中可扰乱微生物群落结构并阻止其他细菌定殖[10]。
中肠是蜜蜂消化道中最大的器官。中肠中3种主要共生菌(即Beta、Firm-5和Gamma-1,简称BFG)只占BFG微生物总数的1%-4%[10]。由于中肠上皮细胞可产生围食膜,围食膜是一种松散的薄膜可以辅助消化,保护上皮细胞以防粗糙的食物粒子[49-50]。中肠上皮细胞可连续生产这种膜状物并在食物通过时脱落,这样可以阻止微生物附着[49]。因此,消化酶和围食膜的存在可以解释中肠中肠道菌比较贫瘠的原因。FISH显微检测结果表明大多数中肠细菌都处于较后的位置,靠近幽门部,表明中肠的部分微生物可能在解剖切割中肠时从回肠转入到中肠后部[10]。
蜜蜂肠道后肠内部有稳定的角质层,后肠分为两个不同的部分,回肠和直肠,每个部分都由不同的微生物群落组成。蜜蜂后肠是由一些简单而高度特异的细菌群落组成[7, 51],它主要包括5个核心成员,普遍存在于世界各地的工蜂肠道中[10]。回肠是一种具有6个纵向折叠的细管,主要由革兰氏阴性细菌控制。这个细菌群落在幽门及中肠、回肠和马氏管的交界处也有发现。回肠中Beta种系型的细菌一层层地粘附在临近的宿主肠道组织上,Gamma-1种系型的细菌厚厚地分布在临近Beta细菌和回肠壁附近。中肠中S. alvi、Firm-5和G. apicola这3种细菌在整个肠道中占总数的1%-4%,尽管中肠比回肠大,但回肠中这3种细菌数量几乎比中肠大两倍。与中肠相比,回肠折叠的内膜具有丰富的附着点,使其有机会接触一些消化而未被吸收的营养物质。回肠的细菌群落相对于肠道壁来说表现为分层次的生物膜[10],邻近宿主组织部位具有Beta种系型生物膜结构,Gamma-1种系型分布在邻近Beta和回肠壁的厚垫上,此外,Firm-5种系型沿着回肠壁周围的小囊中分布[10]。Beta种系型的附着可能有利于随后的细菌种系型定殖,如易于Gamma-1的定殖和附着[10]。此外,其产生的生物薄膜可能产生微梯度(Microgradients) (如营养、氧气和pH),这些可能提供分散的生态位以利于各种底物的利用,类似于白蚁的腹部菌群[52]。
研究表明Firm-5菌群在蜜囊和直肠内占优势,Gilliamella为中肠内的优势菌群,而Snodgrassella为回肠中的优势菌群[10];在幽门部位,F. perrara含量最为丰富(图 1[53])。此外,Lactobacillus和Bifidobacterium类群也主要栖息在直肠中[10, 34]。乳酸杆菌属物种也可以在回肠腔内发现,但在直肠中含量最丰富。直肠的内容物(主要是空的花粉外壁)可为细菌提供营养来源,因此其中栖息着大部分稳定的微生物,占每只蜜蜂总的BFG 16S rRNA基因总数的87%-94%[10]。这3种主要共生菌S. alvi、Firm-5和G. apicola在直肠中的数量占其在每只蜜蜂肠道中总数的87%-94%[10],但这一研究主要基于qPCR的绝对定量研究的结果,并未采用最新的二代测序技术来研究,因此可能并未全面反映出蜜蜂肠道在空间上的定殖特征及规律。总的来说,直肠包含了大多数BFG种系型的16S rRNA基因拷贝,并且还含有一些额外的细菌细胞,这些细菌只与通用真细菌探针杂交而不与特定的BFG探针杂交。这些非BFG细菌细胞大部分代表了除去特定特征微生物后的剩余的种系型(如Alpha-1、Alpha-2.1、Alpha-2.2、Bifido、Firm-4和Gamma-2)[10]。其他非核心成员可以被归类为专门的蜜蜂肠道菌群,它们存在于几乎所有的蜂巢中,但不是每一个个体(如F. perrara)[54]或是在蜂巢或花的环境中发现的细菌,偶尔也会在蜂巢中繁殖。到目前为止,能够使肠道菌群定殖的分子机制尚未阐明,但对于生物膜形成的几种肠道菌群基因已经有了普遍的认识。

对蜜蜂来说,肠道上皮细胞的免疫应答性并不仅限于蜂巢内不同蜜蜂种类所带来的差异,这可能也是由于营养物质摄入或其他压力所产生,偶尔个别成年工蜂的肠道菌群组成有很大的偏差。这种转变通常涉及更大量的机会性环境细菌,包括肠杆菌科成员(例如,Klebsiella、Pantoea、Enterobacter、Serratia和Hafnia)和其他Gammaprotoebacteria[38],这些转变可能反应了菌群的混乱或疾病:在某些方面,它们类似于机会性变形杆菌对人类肠道的侵袭,这种现象是克罗恩病等多种疾病的特征[55],而熊蜂似乎比蜜蜂更容易出现这种转变。在对美国新泽西州的3种本土熊蜂的肠道群落组成的研究中,所有的物种都携带有S. alvi、G. apicola、Firm-4和Firm-5[56]。然而,一些个别蜜蜂包含混乱的微生物群落,具有不稳定的微生物群组成,其特征在于蜜蜂特异性细菌种类少以及高含量的乙酸杆菌科和肠杆菌科环境成员。这些微生物群落在所有3种宿主物种中均有发现,但它们变化多样,并且与真核肠道寄生虫的存在呈正相关[25, 57]。同样,对中国的28种熊蜂物种的16S rRNA基因调查中,肠道微生物群落分为两种不同的组成类型,不同物种以不同频率分布。第一种类型是正常的蜂特异性微生物群落,以S. alvi、G. apicola、Firm-4和Firm-5为代表;另一种是多种肠杆菌科和其他变形菌以及环境乳酸菌占优势。
微生物群落被破坏可能的原因是抗生素的使用,分析美国蜜蜂的特征性肠道微生物群,发现四环素抗性基因频繁出现,这可能是使用抗生素治疗对蜜蜂肠道微生物造成的强选择性,也类似于在人类肠道菌群中起作用的抗生素的选择[58]。研究蜜蜂核心肠道菌群(G. apicola和S. alvi)两个丰富的菌株的系统发育关联性、基因组含量[(G+C)mol%含量、基因组大小、基因数量和CRISPR (Clustered regularly interspaced short palindromic repeats)]和抗生素抗性基因(Antibiotic resistance genes,ARG),结果表明蜜蜂肠道共生亚群可以抵抗长期抗生素治疗,并通过水平基因转移获取地理上不同的抗生素抗性基因来维持功能[59]。在美国,土霉素经常被用于预防和治疗美洲幼虫腐臭病(幼虫芽孢杆菌的感染)[60-63],由于这一点,在蜜蜂肠道中已经鉴定出几种不同的四环素抗性(TCr)基因[64]。相反,不使用土霉素养蜂的国家检测到较少的TCr基因,也不太丰富[64]。
2 宿主的功能和新陈代谢对肠道菌群的影响 2.1 对蜜蜂健康的影响蜜蜂的肠道细菌具有中和饮食毒素、合成营养物质和消化食物的作用。蜜蜂在全球不同地区的贸易无疑导致了传染病的增加,蜜蜂和野生传粉者之间可能会有疾病传播[65]。花粉是蜜蜂食谱中最重要的一份子,体外分解有助于消化花粉,因为蜜蜂和大多数动物本身不能产生果胶酶。在最近的基因组研究的基础上揭示了蜜蜂肠道微生物群落中的代谢活性和细菌间相互作用,研究也发现蜜蜂存在与果蝇类似的免疫系统的信号传导途径和免疫效应特点,但是现在对于这方面的研究仍有不足。
蜜蜂和熊蜂肠道微生物群是相似的,Beta、Gamma-1、Firm-4、Lacto-2、Firm-5和Lacto-1[7, 9, 66]基本存在,但也检测到Alphaproteobacteria、Betaproteobacteria和Gammaproteobacteria的成员[10, 57],在某些熊蜂物种之间,肠道微生物组成和物种丰富度显著不同,并且肠道微生物组成在不同的位置上是一致的,这种地理一致性与内共生菌特异性结合可能揭示出强烈的功能依赖性。对熊蜂肠道微生物群的大规模培养,揭示了一种能够抑制病原体的细菌物种多样性,此外,在熊蜂肠道中首次报道了几种乳酸菌例如Weissella bombi、Fructobacillus tropeaoli、Convivina intestini和Leuconostoc spp.[67]。
蜜蜂和熊蜂对新烟碱的敏感性由CYP9Q亚家族的细胞色素P450决定的,实验证明不是由于对受体的亲和力的差异,而是在于P450的异源代谢[68]。蜜蜂传粉者具有生物化学防御系统,这些系统决定了它们对杀虫剂的敏感性,这些知识可以用来保护蜜蜂的健康。
2.2 蜜蜂肠道菌群在新陈代谢中的作用通过宏基因组学和转录组学能够充分了解分析蜜蜂肠道菌群的潜在功能特性,并对微生物群落中核心菌群进行相关分析,发现均在代谢过程扮演着重要的角色。大多数蜜蜂肠道细菌拥有相对较小的基因组,它们失去了某些核心代谢功能,但编码了生物膜形成和细胞间相互作用的通路[29, 44, 69]。G. apicola和S. alvi都有相对较小的基因组,这与它们是特殊的肠道共生体相一致,S. alvi失去了将碳水化合物转化为能源的能力,因为糖酵解、磷酸戊糖途径和需要将糖转化为丙酮酸的Entner-Doudoroff通路都缺乏关键的酶。但是S. alvi具有吸收羧酸盐的转运蛋白,转运蛋白直接用于三羧酸(Tricarboxylic acid,TCA)循环或在有乳酸的情况下,可以通过乳酸脱氢酶转化为丙酮酸[31]。这些代谢差异也暗示了S. alvi和发酵菌株对不同资源的利用,因为该菌所使用的一些底物(乳酸、醋酸盐和甲酸盐)是通过碳水化合物发酵后得到。Parasaccharibacter apium是一种具有特殊生态位的细菌,虽然不能通过糖和醇氧化生成乙酸,但是却能够适应蜂王浆的条件并可在蜂后肠道内脏中定殖。
蜜蜂收集的食物富含碳水化合物,然而食物中或者花粉分解产生的某些碳水化合物对蜜蜂来说是有毒的。蜜蜂肠道中最主要的发酵菌是革兰氏阴性菌G. apicola和革兰氏阳性乳酸杆菌种系Firm-4和Firm-5[53]。所有的G. apicola菌株均具有较高的16S rRNA基因相似性,但它们与相关的碳水化合物代谢存在广泛的差异。比如一些菌株可以利用甘露糖、阿拉伯糖、木糖或鼠李糖(单糖对蜜蜂有毒性作用)作为它们唯一的碳来源。考虑到它们同时利用葡萄糖、果糖和甘露糖的能力,以及许多菌株分解其他潜在毒性碳水化合物的能力,G. apicola细菌可能在改善饮食耐受性和保持蜜蜂宿主的健康方面具有关键作用[70]。最近对Firm-4和Firm-5基因组的测序揭示了许多涉及摄取糖的磷酸转移酶系统,尤其是乳酸菌Firm-5,蜜蜂相关的双歧杆菌和乳酸杆菌菌株均具有大量功能未知的假定的细胞表面蛋白,其可能参与植物化合物的粘附或降解[31, 71]。
3 小结与展望蜜蜂授粉对于全球生态环境极具重要性。蜜蜂种群的衰退已经引起了人们对影响其健康的潜在因子的关注。研究表明,蜜蜂肠道中的细菌有助于防御入侵的病原体,而这些防御方式目前还不清楚。蜜蜂肠道菌群的特殊性和稳定性的研究是研究病原菌感染蜜蜂的基础[70],目前已经明确日龄[10]、群体[39]和季节[42-43]会对蜜蜂肠道菌群产生影响,但在影响机制方面研究还不足。本土中蜂的肠道菌群受宿主日龄的影响较大,与西方蜜蜂定殖规律不同,而且我国兰州熊蜂定殖规律与中蜂相似[37]。因此开展东、西方蜜蜂肠道菌群的时空定殖规律的对比研究十分必要,可为进一步揭示东、西方蜜蜂肠道菌群定殖的共性及差异提供理论基础。另外,蜜蜂肠道菌群的研究结论主要基于西方蜜蜂,而对我国本土其他蜜蜂属、熊蜂属的相关研究还不够深入。研究表明肠道菌群具有宿主特异性,但这种特异性是如何维持的[52],还需要进一步探究。
蜜蜂肠道作为一个新兴的研究肠道细菌的模型,具有既实用又适合研究共生理论的优势。与人类相比,昆虫的共生菌数量较少,但它们对宿主更重要。本文总结了蜜蜂肠道菌群的种类、定殖规律,并对蜜蜂肠道菌群的影响因素进行了简要介绍。研究蜜蜂肠道菌群的定殖规律,有助于我们了解细菌是如何传递和传播的,同时也为研究肠道细菌的多样化及与宿主的适应进化奠定基础。随着基因组学、转录组学及宏转录组学的进一步发展,为我们更好地研究蜜蜂肠道菌群精细定殖规律提供了技术支持,对深入研究病原菌入侵及与肠道菌群的互作机理及肠道菌群的功能特性奠定了基础。
| [1] |
Kostic AD, Howitt MR, Garrett WS. Exploring host-microbiota interactions in animal models and humans[J]. Genes and Development, 2013, 27(7): 701-718. DOI:10.1101/gad.212522.112 |
| [2] |
Moeller AH, Foerster S, Wilson ML, et al. Social behavior shapes the chimpanzee pan-microbiome[J]. Science Advances, 2016, 2(1): e1500997. DOI:10.1126/sciadv.1500997 |
| [3] |
Calderone NW. Insect pollinated crops, insect pollinators and US agriculture: trend analysis of aggregate data for the period 1992-2009[J]. PLoS One, 2012, 7(5): e37235. DOI:10.1371/journal.pone.0037235 |
| [4] |
Fries I, Chauzat MP, Chen YP, et al. Standard methods for Nosema research[J]. Journal of Apicultural Research, 2013, 52(1): 1-28. |
| [5] |
de Miranda JR, Bailey L, Ball BV, et al. Standard methods for virus research in Apis mellifera[J]. Journal of Apicultural Research, 2013, 52(4): 1-56. |
| [6] |
Koch H, Schmid-Hempel P. Socially transmitted gut microbiota protect bumble bees against an intestinal parasite[J]. Proceedings of the National Academy of Sciences of the United States of America, 2011, 108(48): 19288-19292. DOI:10.1073/pnas.1110474108 |
| [7] |
Martinson VG, Danforth BN, Minckley RL, et al. A simple and distinctive microbiota associated with honey bees and bumble bees[J]. Molecular Ecology, 2011, 20(3): 619-628. DOI:10.1111/j.1365-294X.2010.04959.x |
| [8] |
Sabree ZL, Hansen AK, Moran NA. Independent studies using deep sequencing resolve the same set of core bacterial species dominating gut communities of honey bees[J]. PLoS One, 2012, 7(7): e41250. DOI:10.1371/journal.pone.0041250 |
| [9] |
Moran NA, Hansen AK, Powell JE, et al. Distinctive gut microbiota of honey bees assessed using deep sampling from individual worker bees[J]. PLoS One, 2012, 7(4): e36393. DOI:10.1371/journal.pone.0036393 |
| [10] |
Martinson VG, Moy J, Moran NA. Establishment of characteristic gut bacteria during development of the honeybee worker[J]. Applied and Environmental Microbiology, 2012, 78(8): 2830-2840. DOI:10.1128/AEM.07810-11 |
| [11] |
Li JL, Powell JE, Guo J, et al. Two gut community enterotypes recur in diverse bumblebee species[J]. Current Biology, 2015, 25(15): R652-R653. DOI:10.1016/j.cub.2015.06.031 |
| [12] |
Powell JE, Martinson VG, Urban-Mead K, et al. Routes of acquisition of the gut microbiota of the honey bee Apis mellifera[J]. Applied and Environmental Microbiology, 2014, 80(23): 7378-7387. DOI:10.1128/AEM.01861-14 |
| [13] |
Corby-Harris V, Maes P, Anderson KE. The bacterial communities associated with honey bee (Apis mellifera) foragers[J]. PLoS One, 2014, 9(4): e95056. DOI:10.1371/journal.pone.0095056 |
| [14] |
Kwong WK, Moran NA. Cultivation and characterization of the gut symbionts of honey bees and bumble bees: description of Snodgrassella alvi gen. nov., sp. nov., a member of the family Neisseriaceae of the Betaproteobacteria, and Gilliamella apicola gen. nov., sp. nov., a member of Orbaceae fam. nov., Orbales ord. nov., a sister taxon to the order 'Enterobacteriales' of the Gammaproteobacteria[J]. International Journal of Systematic and Evolutionary Microbiology, 2013, 63: 2008-2018. DOI:10.1099/ijs.0.044875-0 |
| [15] |
Philipp E, Kwong WK, Moran NA. Frischella perrara gen. nov., sp. nov., a Gammaproteobacterium isolated from the gut of the honeybee, Apis mellifera[J]. International Journal of Systematic and Evolutionary Microbiology, 2013, 63(10): 3646-3651. |
| [16] |
Moran NA. Genomics of the honey bee microbiome[J]. Current Opinion in Insect Science, 2015, 10: 22-28. DOI:10.1016/j.cois.2015.04.003 |
| [17] |
Corby-Harris V, Snyder LA, Schwan MR, et al. Origin and effect of Alpha 2. 2 Acetobacteraceae in honey bee larvae and description of Parasaccharibacter apium gen. nov., sp. nov.[J]. Applied Environmental Microbiology, 2014, 80(24): 7460-7472. DOI:10.1128/AEM.02043-14 |
| [18] |
McFrederick QS, Cannone JJ, Gutell RR, et al. Specificity between lactobacilli and hymenopteran hosts is the exception rather than the rule[J]. Applied and Environmental Microbiology, 2013, 79(6): 1803-1812. DOI:10.1128/AEM.03681-12 |
| [19] |
McFrederick QS, Wcislo WT, Hout MC, et al. Host species and developmental stage, but not host social structure, affects bacterial community structure in socially polymorphic bees[J]. FEMS Microbiology Ecology, 2014, 88(2): 398-406. DOI:10.1111/1574-6941.12302 |
| [20] |
McFrederick QS, Wcislo WT, Taylor DR, et al. Environment or kin: whence do bees obtain acidophilic bacteria?[J]. Molecular Ecology, 2012, 21(7): 1754-1768. DOI:10.1111/j.1365-294X.2012.05496.x |
| [21] |
Ahn JH, Hong IP, Bok JI, et al. Pyrosequencing analysis of the bacterial communities in the guts of honey bees Apis cerana and Apis mellifera in Korea[J]. Journal of Microbiology, 2012, 50(5): 735-745. DOI:10.1007/s12275-012-2188-0 |
| [22] |
Koch H, Schmid-Hempel P. Bacterial communities in central European bumblebees: low diversity and high specificity[J]. Microbial Ecology, 2011, 62(1): 121-133. DOI:10.1007/s00248-011-9854-3 |
| [23] |
Babendreier D, Joller D, Romeis J, et al. Bacterial community structures in honeybee intestines and their response to two insecticidal proteins[J]. FEMS Microbiology Ecology, 2007, 59(3): 600-610. DOI:10.1111/j.1574-6941.2006.00249.x |
| [24] |
Lim HC, Chu CC, Seufferheld MJ, et al. Deep sequencing and ecological characterization of gut microbial communities of diverse bumble bee species[J]. PLoS One, 2015, 10(3): e0118566. DOI:10.1371/journal.pone.0118566 |
| [25] |
Martinson VG, Magoc T, Koch H, et al. Genomic features of a bumble bee symbiont reflect its host environment[J]. Applied and Environmental Microbiology, 2014, 80(13): 3793-3803. DOI:10.1128/AEM.00322-14 |
| [26] |
Killer J, Kopečný J, Mrázek J, et al. Bombiscardovia coagulans gen. nov., sp. nov., a new member of the family Bifidobacteriaceae isolated from the digestive tract of bumblebees[J]. Systematic and Applied Microbiology, 2010, 33(7): 359-366. DOI:10.1016/j.syapm.2010.08.002 |
| [27] |
Praet J, Meeus I, Cnockaert M, et al. Bifidobacterium commune sp. nov. isolated from the bumble bee gut[J]. Antonie Van Leeuwenhoek, 2015, 107(5): 1307-1313. DOI:10.1007/s10482-015-0425-3 |
| [28] |
Koch H, Abrol DP, Li JL, et al. Diversity and evolutionary patterns of bacterial gut associates of corbiculate bees[J]. Molecular Ecology, 2013, 22(7): 2028-2044. DOI:10.1111/mec.12209 |
| [29] |
Kwong WK, Engel P, Koch H, et al. Genomics and host specialization of honey bee and bumble bee gut symbionts[J]. Proceedings of the National Academy of Sciences of the United States of America, 2014, 111(31): 11509-11514. DOI:10.1073/pnas.1405838111 |
| [30] |
Kwong WK, Moran NA. Evolution of host specialization in gut microbes: the bee gut as a model[J]. Gut Microbes, 2015, 6(3): 214-220. DOI:10.1080/19490976.2015.1047129 |
| [31] |
Tamarit D, Ellegaard KM, Wikander J, et al. Functionally structured genomes in Lactobacillus kunkeei colonizing the honey crop and food products of honeybees and stingless Bees[J]. Genome Biology and Evolution, 2015, 7(6): 1455-1473. DOI:10.1093/gbe/evv079 |
| [32] |
Guo J. Diversity and influencing factors of gut microbiota in honey bees[D]. Beijing: Doctoral Dissertation of Graduate School of Chinese Academy of Agricultural Sciences, 2015 (in Chinese) 郭军.蜜蜂肠道菌群多样性及其影响因素研究[D].北京: 中国农业科学院研究生院博士学位论文, 2015 http://cdmd.cnki.com.cn/Article/CDMD-82101-1015378973.htm |
| [33] |
Seeley TD. Adaptive significance of the age polyethism schedule in honeybee colonies[J]. Behavioral Ecology and Sociobiology, 1982, 11(4): 287-293. DOI:10.1007/BF00299306 |
| [34] |
Anderson KE, Rodrigues PAP, Mott BM, et al. Ecological succession in the honey bee gut: shift in Lactobacillus strain dominance during early adult development[J]. Microbial Ecology, 2016, 71(4): 1008-1019. DOI:10.1007/s00248-015-0716-2 |
| [35] |
Kapheim KM, Rao VD, Yeoman CJ, et al. Caste-specific differences in hindgut microbial communities of honey bees (Apis mellifera)[J]. PLoS One, 2015, 10(4): e0123911. DOI:10.1371/journal.pone.0123911 |
| [36] |
Guo J, Wu J, Chen YP, et al. Characterization of gut bacteria at different developmental stages of Asian honey bees, Apis cerana[J]. Journal of Invertebrate Pathology, 2015, 127: 110-114. DOI:10.1016/j.jip.2015.03.010 |
| [37] |
Anderson KE, Carroll MJ, Sheehan T, et al. Hive-stored pollen of honey bees: many lines of evidence are consistent with pollen preservation, not nutrient conversion[J]. Molecular Ecology, 2015, 23(23): 5904-5917. |
| [38] |
Tarpy DR, Mattila HR, Newton ILG. Development of the honey bee gut microbiome throughout the queen-rearing process[J]. Applied and Environmental Microbiology, 2015, 81(9): 3182-3191. DOI:10.1128/AEM.00307-15 |
| [39] |
Lee FJ, Rusch DB, Stewart FJ, et al. Saccharide breakdown and fermentation by the honey bee gut microbiome[J]. Environmental Microbiology, 2015, 17(3): 796-815. DOI:10.1111/1462-2920.12526 |
| [40] |
Hroncova Z, Havlik J, Killer J, et al. Variation in honey bee gut microbial diversity affected by ontogenetic stage, age and geographic location[J]. PLoS One, 2015, 10(3): e0118707. DOI:10.1371/journal.pone.0118707 |
| [41] |
Ludvigsen J, Rangberg A, Avershina E, et al. Shifts in the midgut/pyloric microbiota composition within a honey bee apiary throughout a season[J]. Microbes and Environments, 2015, 30(3): 235-244. DOI:10.1264/jsme2.ME15019 |
| [42] |
Mohr KI, Tebbe CC. Diversity and phylotype consistency of bacteria in the guts of three bee species (Apoidea) at an oilseed rape field[J]. Environmental Microbiology, 2006, 8(2): 258-272. DOI:10.1111/j.1462-2920.2005.00893.x |
| [43] |
Ludvigsen J. Seasonal trends in the midgut microbiota of honeybees[D]. Oslo: Master's Thesis of Norwegian University of Life Sciences, 2013
|
| [44] |
Engel P, Moran NA. Functional and evolutionary insights into the simple yet specific gut microbiota of the honey bee from metagenomic analysis[J]. Gut Microbes, 2013, 4(1): 60-65. DOI:10.4161/gmic.22517 |
| [45] |
Sammataro D, Cicero JM. Functional morphology of the honey stomach wall of the European honey bee (Hymenoptera: Apidae)[J]. Annals of the Entomological Society of America, 2010, 103(6): 979-987. DOI:10.1603/AN09111 |
| [46] |
Snodgrass RE. The anatomy of the honey bee[J]. Nature, 1910, 18(2145): 169. |
| [47] |
Èile B, Alsterfjord M, Olofsson TC, et al. Proteins of novel lactic acid bacteria from Apis mellifera mellifera: an insight into the production of known extra-cellular proteins during microbial stress[J]. BMC Microbiology, 2013, 13: 235. DOI:10.1186/1471-2180-13-235 |
| [48] |
Anderson KE, Johansson A, Sheehan TH, et al. Draft genome sequences of two Bifidobacterium sp. from the honey bee (Apis mellifera)[J]. Gut Pathogens, 2013, 5: 42. DOI:10.1186/1757-4749-5-42 |
| [49] |
Tellam RL. The peritrophic matrix[A]//Lehane MJ, Billingsley PF. Biology of the Insect Midgut[M]. Dordrecht: Springer, 1996: 86-114
|
| [50] |
Yue D, Nordhoff M, Wieler LH, et al. Fluorescence in situ hybridization (FISH) analysis of the interactions between honeybee larvae and Paenibacillus larvae, the causative agent of American foulbrood of honeybees (Apis mellifera)[J]. Environmental Microbiology, 2010, 10(6): 1612-1620. |
| [51] |
Kwong WK, Medina LA, Koch H, et al. Dynamic microbiome evolution in social bees[J]. Science Advances, 2017, 3(3): e1600513. DOI:10.1126/sciadv.1600513 |
| [52] |
Brune A, Friedrich M. Microecology of the termite gut: structure and function on a microscale[J]. Current Opinion in Microbiology, 2000, 3(3): 263-269. DOI:10.1016/S1369-5274(00)00087-4 |
| [53] |
Kwong WK, Moran NA. Gut microbial communities of social bees[J]. Nature Reviews Microbiology, 2016, 14(6): 374-384. DOI:10.1038/nrmicro.2016.43 |
| [54] |
Engel P, Bartlett KD, Moran NA. The bacterium Frischella perrara causes scab formation in the gut of its honeybee host[J]. mBio, 2015, 6(3): e00193-15. DOI:10.1128/mBio.00193-15 |
| [55] |
Ilseung C, Blaser MJ. The human microbiome: at the interface of health and disease[J]. Nature Reviews Genetics, 2012, 13(4): 260-270. DOI:10.1038/nrg3182 |
| [56] |
Jones JC, Fruciano C, Hildebrand F, et al. Gut microbiota composition is associated with environmental landscape in honey bees[J]. Ecology and Evolution, 2018, 8(1): 441-451. DOI:10.1002/ece3.3597 |
| [57] |
Cariveau DP, Powell JE, Koch H, et al. Variation in gut microbial communities and its association with pathogen infection in wild bumble bees (Bombus)[J]. The ISME Journal, 2014, 8(12): 2369-2379. DOI:10.1038/ismej.2014.68 |
| [58] |
Blaser MJ. Missing microbes: how the overuse of antibiotics is fueling our modern plagues[J]. Emerging Infectious Diseases, 2014, 20(11): 1961. DOI:10.3201/eid2011.141052 |
| [59] |
Ludvigsen J, Porcellato D, L'Abée-Lund TM, et al. Geographically widespread honeybee-gut symbiont subgroups show locally distinct antibiotic-resistant patterns[J]. Molecular Ecology, 2017, 26(23): 6590-6607. DOI:10.1111/mec.14392 |
| [60] |
Genersch E. American foulbrood in honeybees and its causative agent, Paenibacillus larvae[J]. Journal of Invertebrate Pathology, 2010, 103 Suppl: S10-S19. |
| [61] |
Reybroeck W, Daeseleire E, de Brabander HF, et al. Antimicrobials in beekeeping[J]. Veterinary Microbiology, 2012, 158(1/2): 1-11. |
| [62] |
Spivak M. Preventative antibiotic treatments for honey bee colonies[J]. American Bee Journal, 2000, 140(11): 867-868. |
| [63] |
Thompson HM, Waite RJ, Wilkins S, et al. Effects of European foulbrood treatment regime on oxytetracycline levels in honey extracted from treated honeybee (Apis mellifera) colonies and toxicity to brood[J]. Food Additives & Contaminants, 2005, 22(6): 573-578. |
| [64] |
Tian B, Fadhil NH, Powell JE, et al. Long-term exposure to antibiotics has caused accumulation of resistance determinants in the gut microbiota of honeybees[J]. mBio, 2012, 3(6): e00377-12. DOI:10.1128/mBio.00377-12 |
| [65] |
Regan T, Barnett MW, Laetsch DR, et al. Characterisation of the British honey bee metagenome[J]. Nature Communications, 2018, 9(1): 4995. DOI:10.1038/s41467-018-07426-0 |
| [66] |
Meeus I, Parmentier L, Billiet A, et al. 16S rRNA amplicon sequencing demonstrates that indoor-reared bumblebees (Bombus terrestris) harbor a core subset of bacteria normally associated with the wild host[J]. PLoS One, 2015, 10(4): e0125152. DOI:10.1371/journal.pone.0125152 |
| [67] |
Praet J, Parmentier A, Schmid-Hempel R, et al. Large-scale cultivation of the bumblebee gut microbiota reveals an underestimated bacterial species diversity capable of pathogen inhibition[J]. Environmental Microbiology, 2018, 20(1): 214-227. DOI:10.1111/1462-2920.13973 |
| [68] |
Manjon C, Troczka BJ, Zaworra M, et al. Unravelling the molecular determinants of bee sensitivity to neonicotinoid insecticides[J]. Current Biology, 2018, 28(7): 1137-1143. DOI:10.1016/j.cub.2018.02.045 |
| [69] |
Ellegaard KM, Tamarit D, Javelind E, et al. Extensive intra-phylotype diversity in lactobacilli and bifidobacteria from the honeybee gut[J]. BMC Genomics, 2015, 16: 284. DOI:10.1186/s12864-015-1476-6 |
| [70] |
Zheng H, Nishida A, Kwong WK, et al. Metabolism of toxic sugars by strains of the bee gut symbiont Gilliamella apicola[J]. mBio, 2016, 7(6): e01326-16. DOI:10.1128/mBio.01326-16 |
| [71] |
Kwong WK, Mancenido AL, Moran NA. Genome sequences of Lactobacillus sp. strains wkB8 and wkB10, members of the Firm-5 clade, from honey bee guts[J]. Genome Announcements, 2014, 2(6): e01176-14. DOI:10.1128/genomeA.01176-14 |