 2017, Vol. 44
2017, Vol. 44扩展功能
文章信息
- 吾尔恩·阿合别尔迪, 焦子伟, 江波拉提, 木古丽, 杨晓绒
- OREN Akhberdi, JIAO Zi-Wei, Janbolat, MU Gu-Li, YANG Xiao-Rong
- 高通量测序技术分析新疆新源县过度放牧土壤细菌多样性
- Determination of bacteria diversity of degraded grassland in Xinyuan county by high-throughput sequencing technology
- 微生物学通报, 2017, 44(3): 545-553
- Microbiology China, 2017, 44(3): 545-553
- DOI: 10.13344/j.microbiol.china.160627
-
文章历史
- 收稿日期: 2016-08-31
- 接受日期: 2016-11-07
- 优先数字出版日期(www.cnki.net): 2016-12-19






2. 南开大学生命科学学院 微生物系 天津 300071
2. Department of Microbiology, college of life Sciences, Nankai University, Tianjin 300071, China
近年来由于对自然资源的掠夺式开发,使得生物多样性遭到严重破坏,生态系统渐趋波动。而土壤微生物作为稳定生态系统、监测土壤质量变化的敏感指标,其多样性研究在评价生态系统、维护生态平衡中发挥了巨大作用,因此越来越多的学者将目光投向土壤微生物多样性的研究和保护[1]。
土壤微生物不仅是土壤的重要组成部分,更是土壤养分循环的主要推动者,土壤微生物群落的变化在一定程度上可以反映土壤质量的变化趋势[2-3]。在土壤微生物中,以细菌的种类和数量最多[4],细菌在土壤营养元素循环、有机质的形成与分解、土壤结构成分的形成、生态环境的改善、植物的生长发育和作物病虫害防治等方面均起着极其重要的作用[5],因此细菌多样性的研究受到广泛关注。生物固氮是土壤中有效性氮素的重要来源[6-7],构成全球氮循环的中心环节,全球每年由微生物介导的固氮达1.5×1013 mol。固氮微生物是除人为施肥因素外土壤氮素来源的主要贡献者,其群落结构组成对土壤氮素固定及维持氮素循环平衡具有重要意义。
新疆伊犁河谷是新疆乃至全国重要的畜牧业基地,畜牧业生产主要是以家庭牧场为单元的生产经营方式。随着牲畜数量的不断增加、草原面积缩小、冷季饲草不足,使得草畜不平衡十分突出,伊犁河谷天然草地退化面积221.07万hm2,占总面积的64.65%,占可利用面积的71.21%[8]。本研究以伊犁新源县退化草原土壤为研究对象,采用Illumina HiSeq 2500高通量测序技术,研究土壤细菌的多样性,并对固氮细菌的种类进行分析,为恢复退化草原提供一定依据。
1 材料与方法 1.1 样品采集6个退化土样采自新疆新源县种羊场及其周围地区 (43°33′N,82°41′E−82°58′E),海拔750 m−850 m。第一样品组TJ.1.1、TJ.1.2、TJ.2采自于过度放牧区,而另一组样品WJ.1.1、WJ.1.2、WJ.2采自于近3年受保护的适度放牧草原,之前也是过度放牧区,适度放牧与过度放牧样地由栅栏隔离,如图 1所示。每一样地多点采样法采集0−20 cm表层混合样,带回实验室后去杂过2 mm钢筛,充分混匀装袋备用。

|
| 图 1 适度放牧区和过度放牧区 Figure 1 The overgrazing and suitable grazing grasslands |
|
|
土壤DNA提取试剂盒,MOBIO公司;Phusion® High-Fidelity PCR Master Mix with GC Buffer高保真酶,New England Biolabs公司。Master Cycler gradient PCR仪,Eppendorf公司;Neofuge 23冷冻离心机,HEAL立新仪器公司;ChemiDocTM Xrs凝胶成像仪,Bio-Rad公司。
1.3 土壤微生物基因组DNA的提取DNA的提取采用美国 (MOBIO Power Soil DNA Isolation Kit) 强力土壤DNA提取试剂盒。按照说明书的提取步骤进行。将提取得到的土壤微生物总DNA溶解于100 µL去离子水,取5 µL的DNA用1.0%的琼脂糖凝胶电泳检测 (0.5 ×TAE缓冲液),分析DNA的完整性和相对浓度。
1.4 土壤细菌16S rRNA基因PCR扩增以稀释后的基因组DNA为模板,使用带Barcode的16S rRNA基因V4区特异引物515F (5′-GTTTCGGTGCCAGCMGCCGCGGTAA-3′) 和806R (5′-GCCAATGGACTACHVGGGTWTCTAAT-5′),高效和高保真酶进行PCR扩增,反应体系参照说明书,反应条件:98 ℃ 2 min;98 ℃ 10 s,50 ℃ 30 s,72 ℃ 30 s,30个循环;72 ℃ 10 min。
1.5 土壤细菌16S rRNA基因测序土壤基因组DNA送至北京诺禾致源生物信息科技有限公司进行Illumina HiSeq 2500高通量测序。
1.6 数据分析利用Uparse[9]软件对所有样品的全部有效序列 (Effective tags) 进行聚类,默认以97%的一致性 (Identity) 将序列聚类成为OTUs (Operational taxonomic units),同时选取OTUs的代表性序列,依据其算法原则,筛选OTUs中出现频数最高的序列作为OTUs的代表序列。对OTUs代表序列进行物种注释,用Mothur软件与SILVA[10]的SSUrRNA和GreenGene数据库[11]进行物种注释分析 (设定阈值为0.8−1.0),获得分类学信息用Mothur软件包以97%为划定阈值对16S rRNA基因序列划分操作分类单元 (OTU),并构建稀释曲线[12]。
利用QIIME软件计算样品包括Chao1指数和Shannon指数的Alpha多样性值[13]。其中群落丰富度用Chao1指数描述,其值越高表明群落物种的丰富度越高。而Shannon指数可以反映样品的多样性程度,其值越高表明群落物种的多样性越高。
Chao1丰富度指数:

|
其中,Sobs为观测到的OTU数,F1为只有一条序列的OTU数目,F2为只有两条序列的OTU数目。
Shannon多样性指数:
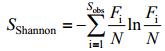
|
其中,Sobs为观测到的OTU数,Fi为含有i条序列的OTU数目,N为所有序列数目。
2 结果与分析 2.1 测序结果质量分析通过对6个样品测序,原始序列条数为432 396,过滤掉低质量的序列后,有效序列的总数为396 578。根据Barcode标签进行样品序列拆分,再对初始序列进行去冗余处理后,获得16S rRNA基因Unique reads,并将其聚类为用于物种分类的OTU,统计得到各个样品在不同OTU中的丰度信息,6个样品产生1 990−2 193个OTU,有效序列比例均大于89%,各样品的相关数据如表 1所示。
| 样品名称 Sample name |
原始序列数量 Raw tags |
有效序列数量 Effective tags |
有效序列比例 Effective (%) |
OTU数量 Number of OTU |
| TJ.1.1 | 63 921 | 57 694 | 90.26 | 1 990 |
| WJ.1.1 | 67 546 | 62 506 | 92.54 | 2 193 |
| TJ.1.2 | 69 689 | 64 195 | 92.12 | 2 156 |
| WJ.1.2 | 68 695 | 61 793 | 89.95 | 2 139 |
| TJ.2 | 85 714 | 80 750 | 94.21 | 2 232 |
| WJ.2 | 76 831 | 69 640 | 90.64 | 2 192 |
稀释曲线 (Rarefaction curve) 反映了样品的取样深度,可以用来评价测序量是否足以覆盖所有类群。从稀释曲线 (图 2) 中可知,序列数量到5 000时各样品稀释曲线均基本趋于平缓,说明取样基本合理,真实环境中细菌群落结构的置信度较高,能够比较真实地反映土壤样本的细菌群落。

|
| 图 2 相似度为0.97条件下各土壤样品的稀释曲线 Figure 2 Rarefaction curves of each soil sample at cutoff level of 3% |
|
|
如表 2所示,Chao1和Shannon指数在适度放牧土壤样品中的平均值分别为2 139.10和8.88,而在过度放牧土样中分别为2 097.45和8.79,说明适度放牧草原土壤中细菌多样性和丰度均有所提高。
| 样地 Sampling site |
Ace指数 Ace index |
Chao1指数 Chao1 index |
Shannon指数 Shannon index |
Simpson指数 Simpson index |
| TJ.1.1 | 1 950.39 | 1 969.28 | 8.780 | 0.995 |
| TJ.1.2 | 2 145.40 | 2 132.68 | 8.860 | 0.995 |
| TJ.2 | 2 253.97 | 2 190.40 | 8.750 | 0.993 |
| WJ.1.1 | 2 188.21 | 2 158.77 | 8.910 | 0.995 |
| WJ.1.2 | 2 118.47 | 2 111.81 | 8.837 | 0.994 |
| WJ.2 | 2 161.57 | 2 146.70 | 8.910 | 0.994 |
在0.97的相似度下,得到了每个样品的OTU个数,利用R Version 2.15.3软件绘出花瓣图,结果可以展示多样品相同和各自特有OTU数目,直观展示样品间OTU的重叠情况。结合OTU所代表的物种,可以找出不同环境中的核心微生物。如图 3所示,6个样品共有的OTU的数量为1 040,而每个样品特有的OTU数量较低。样品TJ.2特有OTU在6个样品中最多,占样品总OTU数2 232的12.4%,而样品TJ.1.2占样品总OTU数的1.3%最低。

|
| 图 3 6个样品中细菌多样性的相关性分析 Figure 3 The similarity analysis of bacteria diversity among six samples |
|
|
6个土壤样品中共检测到细菌门35个,其中丰度较高的前10个门的种类分别为放线菌门 (Actinobacteria)、变形菌门 (Proteobacteria)、厚壁菌门 (Firmicutes)、芽单胞菌门 (Gemmatimonadetes)、拟杆菌门 (Bacteroidetes)、酸杆菌门 (Acidobacteria)、奇古菌门 (Thaumarchaeota)、疣微菌门 (Verrucomicrobia)、浮霉菌门 (Planctomycetes)、绿弯菌门 (Chloroflexi),所占的比例如图 4所示。

|
| 图 4 各土壤样品在门分类水平上细菌类群比较 Figure 4 Comparison of bacteria groups in each soil sample at phylum level |
|
|
其中优势类群为Proteobacteria和Actinobacteria,分别平均比率为31.6%和30.9%,其次是Acidobacteria、Gemmatimonadetes,分别为10.5%和8.9%,其他6个门丰度较低,均小于5.0%,未被分类的细菌占0.4%。
在科的水平上总共220个类群被分类,其中Phingomonadaceae和Gemmatimonadaceae从测序总数中占的比率最高,其次是Rubrobacteriaceae、Geodermatophilaceae、Chitinophagaceae、RB41、Bacillaceae、Solirubrobacteraceae、Micromonosporaceae、Pseudonocardiaceae等,未被分类的细菌占31%,见图 5。

|
| 图 5 各土壤样品在科分类水平上细菌类群比较 Figure 5 Comparison of bacteria groups in each soil sample at family level |
|
|
在属的水平上共有329个类群被分类,其中平均丰度大于1%的有Sphingomonas、Rubrobacter、Blastococcus、Solirubrobacter、Blastocatella、Halomonas、Bacillus、Gemmatimonas、Microvirga等9个属,未知菌类占56.1%,见图 6。科属水平上未知种类占很大比例。

|
| 图 6 各土壤样品在属分类水平上细菌类群比较 Figure 6 Comparison of bacteria groups in each soil sample at genus level |
|
|
适度放牧和过度放牧土壤中一些细菌类群的丰度变化比较大。我们用相对平均偏差,更直观地表达过度和适度放牧土样组间的细菌群落相对丰度的差异。如表 3所示,过度放牧土壤中的Planctomycetes、Verrucomicrobia、Acidobacteria、Chloroflexi、Bacteroidetes等5个门相对丰度低于适度放牧土壤,其中较明显的类群为Planctomycetes、Verrucomicrobia、Acidobacteria,其相对平均偏差分别为45.8%、22.4%和11.4%。Planctomycetes参与多种环境的元素循环,尤其是其中部分种属通过厌氧氨氧化作用参与氮素循环[14]。Verrucomicrobia和Acidobacteria中可培养的种类很少,因此人们对它们生态和代谢作用的认识也甚少,但是它们在自然界中广泛而大量的分布,暗示其在生态系统中的重要作用[15-17]。
| 门 Phylum |
适度放牧土样组中的相对丰度 The relative abundance in overgrazing soil sample group |
过度放牧土样组中的比率 The relative abundance in suitable grazing soil sample group |
两组间的相对平均偏差 Average relative deviation (%) |
| Proteobacteria | 30.1 | 33.1 | 4.7 |
| Actinobacteria | 30.2 | 31.6 | 2.3 |
| Acidobacteria | 11.7 | 9.3 | 11.4 |
| Gemmatimonadetes | 8.6 | 9.1 | 2.8 |
| Bacteroidetes | 5.6 | 5.3 | 2.8 |
| Firmicutes | 3.0 | 3.6 | 9.1 |
| Planctomycetes | 3.5 | 1.3 | 45.8 |
| Verrucomicrobia | 3.0 | 1.9 | 22.4 |
| Thaumarchaeota | 1.2 | 1.9 | 22.6 |
| Chloroflexi | 1.2 | 1.1 | 4.3 |
| Others | 1.9 | 1.8 | 2.7 |
在过度放牧土壤中Thaumarchaeota、Firmicutes、Proteobacteria、Gemmatimonadetes、Actinobacteria等5个门的相对丰度大于适度放牧土壤。其中变化较明显的为Thaumarchaeota,其相对平均偏差为22.6%,这可能与Thaumarchaeota属于具有适应极端环境特性的古细菌有关。Thaumarchaeota是一类通过催化氨氧化获取能量的自养生长古菌,在一些自然生态系统的硝化过程中起着主导作用[18]。
2.5 固氮细菌的分析在属的水平上分析各样品中可能具有固氮能力的细菌 (表 4)。6个土样中自生固氮菌按总测序数依次为Arthrobacter > Methylobacterium > Campylobacter > Desulfovibrio > Propionibacterium。Arthrobacter测序总数最高,达1 078 (占已分类固氮细菌总测序数的33.6%),而该属在种的水平上被分类的有Arthrobacter oxydans,其测序数为860;第二者是总数为706的Methylobacterium(占总测序数22%),该属中仅有4个拷贝的Methylobacterium radiotolerans被注释。6个土样中根瘤固氮菌按总测序数依次为:Bradyrhizobium > Rhizobacter > Mesorhizobium > Rhizobium。测序数较多类群为Bradyrhizobium,该属总测序数636 (占总测序数19.8%) 均被分类为Bradyrhizobium elkanii一个种;Rhizobacter(占总测序数13.4%) 中未发现被注释的种。Bradyrhizobium和Mesorhizobium所有测序数分别被分类为两个种,即Bradyrhizobium elkanii和Mesorhizobium mediterraneum;而Rhizobium属354测序数 (占总测序数11%) 均被分类为Rhizobium larrymoorei一个种。
| 属genus | TJ.1.1 | TJ.1.2 | TJ.2 | WJ.1.1 | WJ.1.2 | WJ.2 | 总共Total |
| 节杆菌属Arthrobacter | 202 | 158 | 229 | 82 | 119 | 288 | 1 078 |
| 甲基杆菌属Methylobacterium | 70 | 338 | 37 | 72 | 63 | 126 | 706 |
| 慢生根瘤菌属Bradyrhizobium | 129 | 215 | 72 | 92 | 43 | 85 | 636 |
| 根瘤杆菌属Rhizobacter | 86 | 58 | 25 | 26 | 147 | 89 | 431 |
| 弯曲杆菌属Campylobacter | 0 | 338 | 16 | 0 | 0 | 0 | 354 |
| 总共Total | 487 | 1 107 | 379 | 272 | 372 | 588 | 205 |
不同学者采用的多样性指数不同,其研究结果存在较大差异,目前应用较广的是丰富度、Shannon指数和均匀度,这3个参数基本代表了土壤微生物群落多样性的特征。土壤微生物群落功能多样性是反映该地区土壤生态系统稳定性的重要指示因子。要较为理想地恢复弃耕地土壤生态系统、建立一个高质量的健康土壤生态应该具有良好的生物活性状况和稳定的微生物种群组成,不仅要恢复地上部植被,还应考虑恢复地下部土壤微生物多样性[19]。
(1) 本研究利用Illumina HiSeq 2500高通量测序技术分析了新源县退化草原土壤的细菌群落结构,研究结果为恢复土壤质量方面提供了理论依据。通过对16S rRNA基因的V4区测序分析,检测了退化草原共6个土壤样品微生物总体群落结构。测序共获得396 578条有效序列,6个样品在97%的相似度水平上可被分为1 990−2 193种水平分类的细菌 (OTU)。适度放牧土样的Chao1和Shannon指数平均值均大于过度放牧土样,说明原来过度放牧区中开始适度放牧之后土壤中细菌的数量和种类都提高。通过与SSUrRNA数据库进行比对,6个土样中的细菌可被分为35个门水平上的类群,优势菌群均依次为Actinobacteria > Proteobacteria > Firmicutes > Gemmatimonadetes > Bacteroidetes > Acidobacteria > Thaumarchaeota > Verrucomicrobia > Planctomycetes > Chloroflexi,其中前4个类群占的比例达到82%。科的水平上共分布于220类群,其中Phingomonadaceae和Gemmatimonadaceae为优势菌,而未被分类细菌平均比例值达到31%。在属的水平上共分布于329个类群,Sphingomonas和Rubrobacter的丰度最高,未被分类细菌平均比例达到56.1%。以上数据说明,虽然退化草原土壤细菌多样性非常丰富,但是优势类群占的比例相当高,许多种类丰度特别低,我们可以推测土壤中很多种类因为不适应正处于的环境条件,数量逐渐减少,趋向灭绝。生态系统中可能具有重要作用的Planctomycetes、Verrucomicrobia、Acidobacteria等类群在适度放牧土壤中的相对丰度大于过度放牧土壤。特别提议的是,因为这些样地附近没有未退化草原,所以现在还不能确定微生物数量和种类的丰度与植被、土壤质量的直接关系。
研究表明细菌内往往存在16S rRNA基因的多拷贝,且基因组内各拷贝之间可能存在异质性,因而基于16S rRNA基因分析菌群多样性时,高估的发生是不可避免的[20]。Sun等发现16S r RNA基因不同区域存在不同程度的异质性,利用不同区域的序列进行多样性分析会导致不同程度的高估[21]。用引物515F/926F扩增的V4−V5区域使高估程度减少至最低 (约3%)。本文采用515F/806R引物对细菌16S rRNA基因进行PCR扩增,该引物扩增平均长度为253 bp片段就在V4区域,因此尽可能地降低了16S rRNA基因在基因组内的多样性引起的OTU数量的高估。
(2) 新型微生物肥料由一种或数种有益微生物活细胞制备而成的肥料。微生物肥料主要靠它含有的大量有益微生物的生命活动来完成,对作物增产和调控植株的生长,改善品质等方面起到重要作用。根瘤菌剂和固氮菌剂是最重要的微生物肥料。本试验对固氮细菌的多样性做了初步的分析。固氮细菌生长繁殖和固氮能力受pH值、酸碱度、盐含量、湿度、温度等很多环境因素的影响,因此摸索适合生长于某地区土壤的菌种是及其重要的。6个土样中自生固氮菌属的分类按总测序数依次为Arthrobacter > Methylobacterium > Campylobacter > Desulfovibrio > Propionibacterium,而根瘤固氮细菌中属的分类依次为Bradyrhizobium > Rhizobacter > Mesorhizobium > Rhizobium,总共找到9个属分类水平上的固氮菌类群。固氮细菌在种的水平上有以下类群:Arthrobacter oxydans、Bradyrhizobium elkanii、Mesorhizobium mediterraneum、Rhizobium larrymoorei、Clostridium beijerinckii。6个退化土样中Arthrobacter、Methylobacterium等2个自生固氮菌和Bradyrhizobium、Rhizobacter等2个根瘤菌的丰度最高,说明这几个种类适合生长于退化草原,恢复土壤研究中可以作为候选微生物肥料。
4 展望当前因过度放牧导致的草原退化现象在新疆伊犁等地区比较严重,而新源县平原放牧草原较广阔,草原退化较为严重,因此我们选择该地区6个样地,每个样地距离较远,几千米至10多千米。我们本来想在每个采样地点附近同时采集未退化草原土壤的样品,与退化土壤做比较,让人遗憾的是这些样地附近没有未退化草原。但近年来许多牧场均通过围栏方式适度放牧。我们相信不久的将来土壤活性以及植被等肯定会有所好转,那时对同一样地土壤做同样分析,与该试验结果做比较,找出土壤细菌丰度和多样性与土壤质量之间的关系。另外,以后将进一步对所分类到的固氮细菌进行针对性地分离纯化并鉴定,利用该菌株制造微生物肥料,用于退化草原土壤当中,观察植被恢复情况。对恢复土壤质量的研究领域提供理论数据。
| [1] | Zhang W, Wei HL, Gao HW, et al. Advances of studies on soil microbial diversity and environmental impact factors[J]. Chinese Journal of Ecology, 2005, 24(1) : 48–52. (in Chinese) 张薇, 魏海雷, 高洪文, 等. 土壤微生物多样性及其环境影响因子研究进展[J]. 生态学杂志, 2005, 24(1) : 48–52. |
| [2] | Li Z, Yuan Y, Ma L, et al. Effects of different rotations on the amount and distribution of soil microorganisms in Mudanjiang tobacco-cropping areas[J]. Journal of Northeast Forestry University, 2010, 38(7) : 96–99. (in Chinese) 李喆, 元野, 马力, 等. 不同轮作方式对牡丹江地区烟田土壤微生物数量及分布的影响[J]. 东北林业大学学报, 2010, 38(7) : 96–99. |
| [3] | Paul EA, Clark FE. Soil Microbiology and Biochemistry[M]. Translated by Gu ZL, Li ZG and Lin XG. Beijing: Scientific and Technical Documentation Press, 1993: 102-273(in Chinese) 波尔EA, 克拉克FE. 土壤微生物学与生物化学[M]. 顾宗濂, 李振高, 林先贵, 译. 北京: 科学技术文献出版社, 1993: 102-273 |
| [4] | Alexander M. Introduction to soil microbiology[J]. Soil Science, 1978, 125(5) : 331. |
| [5] | Kennedy AC. Bacterial diversity in agroecosystems[J]. Agriculture, Ecosystems & Environment, 1999, 74(1/3) : 65–76. |
| [6] | Zou YK, Zhang JN, Yang DL, et al. Effects of different land use patterns on nifH genetic diversity of soil nitrogen-fixing microbial communities in Leymus chinensis steppe[J]. Acta Ecologica Sinica, 2011, 31(3) : 150–156. DOI:10.1016/j.chnaes.2011.03.004 |
| [7] | Dong ZX, Sun B, Yin SX, et al. Impacts of climate and cropping on community diversity of diazotrophs in Pachic udic argiboroll and fluventic ustochrept[J]. Acta Pedologica Sinica, 2012, 49(1) : 130–138. (in Chinese) 董志新, 孙波, 殷士学, 等. 气候条件和作物对黑土和潮土固氮微生物群落多样性的影响[J]. 土壤学报, 2012, 49(1) : 130–138. |
| [8] | Fan TW, Yan K, Zhao DL, et al. Study of forage-livestock balance optimization model of semi-settled family ranch in Yili river valley[J]. Prataculture & Animal Husbandry, 2011(6) : 8–13. (in Chinese) 范天文, 闫凯, 赵德亮, 等. 伊犁河谷半定居家庭牧场草畜平衡优化模式研究[J]. 草业与畜牧, 2011(6) : 8–13. |
| [9] | Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads[J]. Nature Methods, 2013, 10(10) : 996–998. DOI:10.1038/nmeth.2604 |
| [10] | Wang Q, Garrity GM, Tiedje JM, et al. Na ve bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy[J]. Applied and Environmental Microbiology, 2007, 73(16) : 5261–5267. DOI:10.1128/AEM.00062-07 |
| [11] | Quast C, Pruesse E, Yilmaz P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools[J]. Nucleic Acids Research, 2013, 41(D1). |
| [12] | Schloss PD, Westcott SL, Ryabin T, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities[J]. Applied and Environmental Microbiology, 2009, 75(23) : 7537–7541. DOI:10.1128/AEM.01541-09 |
| [13] | Kemp PF, Aller JY. Bacterial diversity in aquatic and other environments: what 16S rDNA libraries can tell us[J]. FEMS Microbiology Ecology, 2004, 47(2) : 161–177. DOI:10.1016/S0168-6496(03)00257-5 |
| [14] | Huang PB, Jiao NZ, Feng J, et al. Research progress on Planctomycetes' diversity and ecological function in marine environments[J]. Microbiology China, 2014, 41(9) : 1891–1902. (in Chinese) 黄佩蓓, 焦念志, 冯洁, 等. 海洋浮霉状菌多样性与生态学功能研究进展[J]. 微生物学通报, 2014, 41(9) : 1891–1902. |
| [15] | Zheng Y, Zheng YM, Zhang LM, et al. Advances in thermoacidophilic methanotrophs from extreme environments[J]. Acta Ecologica Sinica, 2009, 29(7) : 3864–3871. (in Chinese) 郑勇, 郑袁明, 张丽梅, 等. 极端环境下嗜热酸甲烷营养细菌研究进展[J]. 生态学报, 2009, 29(7) : 3864–3871. |
| [16] | Quaiser A, Ochsenreiter T, Lanz C, et al. Acidobacteria form a coherent but highly diverse group within the bacterial domain: evidence from environmental genomics[J]. Molecular Microbiology, 2003, 50(2) : 563–575. DOI:10.1046/j.1365-2958.2003.03707.x |
| [17] | Eichorst SA, Breznak JA, Schmidt TM. Isolation and characterization of soil bacteria that define Terriglobus gen. nov., in the Phylum Acidobacteria[J]. Applied and Environmental Microbiology, 2007, 73(8) : 2708–2717. DOI:10.1128/AEM.02140-06 |
| [18] | Zhang LM, He JZ. A novel archaeal phylum: Thaumarchaeota-a review[J]. Acta Microbiologica Sinica, 2012, 52(4) : 411–421. (in Chinese) 张丽梅, 贺纪正. 一个新的古菌类群--奇古菌门 (Thaumarchaeota)[J]. 微生物学报, 2012, 52(4) : 411–421. |
| [19] | Qin YY, Li JH, Wang G, et al. Effects of sowing legume species on functional diversity of soil microbial communities in abandoned fields[J]. Journal of Lanzhou University (Natural Sciences), 2009, 45(3) : 55–60. (in Chinese) 秦燕燕, 李金花, 王刚, 等. 添加豆科植物对弃耕地土壤微生物多样性的影响[J]. 兰州大学学报:自然科学版, 2009, 45(3) : 55–60. |
| [20] | Pei AY, Oberdorf WE, Nossa CW, et al. Diversity of 16S rRNA genes within individual prokaryotic genomes[J]. Applied and Environmental Microbiology, 2010, 76(12) : 3886–3897. DOI:10.1128/AEM.02953-09 |
| [21] | Sun DL, Jiang X, Wu QL, et al. Intragenomic heterogeneity of 16S rRNA genes causes overestimation of prokaryotic diversity[J]. Applied and Environmental Microbiology, 2013, 79(19) : 5962–5969. DOI:10.1128/AEM.01282-13 |