 2019, Vol. 35
2019, Vol. 35中国科学院微生物研究所、中国微生物学会主办
文章信息
- 刘炳辉, 马洁, 朱云平
- Liu Binghui, Ma Jie, Zhu Yunping
- 细胞外基质蛋白质预测工具研究进展
- Advances in the research of extracellular matrix protein prediction tools
- 生物工程学报, 2019, 35(9): 1571-1580
- Chinese Journal of Biotechnology, 2019, 35(9): 1571-1580
- 10.13345/j.cjb.190043
-
文章历史
- Received: January 23, 2019
- Accepted: March 4, 2019


引用本文

|



2. 国家蛋白质科学中心 (北京) 北京蛋白质组研究中心 蛋白质组学国家重点实验室,北京 102206
2. State Key Laboratory of Proteomics, Beijing Proteome Research Center, National Center for Protein Sciences (Beijing), Beijing 102206, China
多细胞生物的组织由细胞及细胞分泌的大分子网络组成,这些大分子网络就是细胞外基质(Extracellular matrix,ECM)[1]。ECM蛋白质包括胶原、糖蛋白、蛋白聚糖、粘连蛋白、参与ECM形成和重塑的酶以及其他因子[2]。ECM蛋白质在细胞的形成、分化、增殖、生存、极性和迁移中起着至关重要的作用[3-4],其调节异常易导致马凡综合征、成骨不全、软骨发育异常和癌症等诸多疾病的发生[5-8]。因此鉴定细胞外基质蛋白质的组成对于深入理解其功能以及为相关疾病提供有效的治疗靶标具有重要作用。
通过实验鉴定蛋白质的细胞外基质定位需要耗费大量的时间和人力,因此,目前大量蛋白质的定位尚未得到实验验证。为解决这个问题,2012年Naba等[9]通过基于蛋白质功能结构域的半经验方法构建了理论的ECM蛋白质参考数据集——Matrisome,并在2016年发布了更新的2.0版本;同时他们通过对公开的ECM质谱实验数据集进行统一分析并与Matrisome理论列表比较,得到了人类和小鼠的首个ECM草图(ECM Atlas)[1]。ECM Atlas中已有结果显示Matrisome与实验数据的重合度并不高,同时其他研究人员开展的不同物种或者不同组织的ECM蛋白质组研究也证实了类似的结果[10-13]。Naba等认为造成这一结果的原因是多方面的,而基于结构域半经验预测方法本身的局限性可能就是其主要原因之一[1]。
不同于Naba等采用半经验的方法来判定ECM蛋白质,更多的研究者通过机器学习模型发展了一系列ECM蛋白质预测工具,包括:ECMPP[14]、EcmPred[15]、PECM[16]、IECMP[17]、ECMP-HybKNN[18]、BAMORF[19]和TargetECMP[20]等。这些工具都基于“标准数据集选取——特征提取——特征筛选——学习分类——模型性能评估”这一主流生物信息学预测工具流程进行构建,可以实现ECM蛋白质的自动预测,不同的工具分别针对流程中的不同部分进行了创新,由此推动了ECM蛋白质预测工具的不断发展。
文中以基于机器学习模型发展的ECM蛋白质预测工具为主,首先阐述这一类工具构建的基本流程,然后以工具为单位总结已有ECM蛋白质预测工具的研究成果,最后讨论ECM蛋白质预测工具普遍面临的问题和可能的优化方法。
1 ECM蛋白质预测工具构建的基本流程ECM蛋白质预测工具构建的基本流程如图 1所示,可以概括为5个主要步骤:Ⅰ)金标准数据集的构建,包括确定为ECM蛋白质的阳性数据集和确定为非ECM (non-ECM)蛋白质的阴性数据集;Ⅱ)特征提取,将每一个蛋白质序列表示为一个特征向量,特征向量由与ECM蛋白质预测相关的特征组成;Ⅲ)特征筛选,通过特征重要性打分和增量特征选择挑选出最优特征子集,以消除冗余特征带来的噪音;Ⅳ)学习分类,运用随机森林、支持向量机等机器学习算法对数据集进行训练和建模;Ⅴ)模型性能评估,计算模型的敏感度、特异度和准确率等指标以评估模型的性能。

|
| 图 1 细胞外基质蛋白质预测工具构建的基本分析流程 Fig. 1 Overview of the analysis pipeline of ECM protein prediction tools. |
| |
根据已有实验或者数据库注释,选取确定的ECM蛋白质和non-ECM蛋白质分别构成模型训练的阳性数据集和阴性数据集。数据集的选取是构建ECM蛋白质预测工具的基础,从根本上决定了预测工具性能的优劣,因此应尽量保证数据集中ECM和non-ECM蛋白质的准确性。
除ECMPP之外[14],目前已发展的ECM蛋白质预测工具所采用的标准数据集均来自于Kandaswamy等2013年开发EcmPred时所构建的数据集:选取Swiss-Prot数据库release 67版本中的17 233个多细胞动物的分泌蛋白质作为初始数据集,其中1 103个含有ECM注释信息的认定为ECM蛋白质,作为阳性数据集,剩余16 130个不包含ECM注释的蛋白质作为阴性数据集,然后对数据去冗余以保证蛋白质序列两两之间的相似度不超过70%。最后得到由445个ECM蛋白质构成的阳性数据集和4 187个non-ECM蛋白质构成的阴性数据集[15]。
1.2 特征提取在发展蛋白质属性预测方法的计算过程中,蛋白质序列往往被表示为一个特征向量,该向量可以反映出序列与预期目标的内在相关性[21]。发展有效的特征提取方法甚至能比改进分类器实现更高的预测精度[22]。因此,为蛋白质提取精准的特征向量是预测成功的关键步骤。有研究者指出单一特征不能很好地保留足够多的区分信息[23],所以目前绝大部分ECM蛋白质预测工具都提取了多种特征。其中一些代表性的特征得到了大部分研究人员的青睐。
伪氨基酸组成(Pseudo amino acid composition,PseAAC)和二肽组成(Dipeptide composition,DPC)均为基于序列组成信息的特征,可以同时反映蛋白质序列的氨基酸组成信息和氨基酸之间的顺序信息[24-25],在ECM蛋白质预测工具中得到了广泛应用[17-19]。
蛋白质的结构和功能很大程度上由其基本单元氨基酸的理化性质(Physicochemical properties,PP)所定义。经过大量的实验和理论研究,研究者们用氨基酸指数(Amino acid index,AAIndex)来表示每一种氨基酸的理化性质[26]。理化性质已被广泛应用于蛋白质亚细胞定位的研究,目前大多数的ECM蛋白质预测工具也把基于理化性质的特征作为提取的重要特征之一[15-17, 19]。
氨基酸残基的改变、插入和删除等序列变化伴随着蛋白质演变的全部过程[27]。经过漫长的时间,这些累积的进化会慢慢消除初始蛋白和最终蛋白之间的相似性,但是和蛋白质基本属性相关的一些关键性残基倾向于一直保持稳定,表现为进化的保守性。进化保守性通常反映了重要的生物学功能[28]。因此基于进化信息的特征对于蛋白质结构和功能的刻画至关重要[29],在ECM蛋白质预测工具中也得到了广泛的应用[14, 16-17, 20]。位置特异性得分矩阵(Position specific scoring matrix,PSSM)能够很好地反映蛋白质进化信息,如下所示,PSSM由20×L个元素组成,L代表蛋白质序列的长度[30],Ei, j代表在进化过程中序列第i位的氨基酸突变形成氨基酸j的得分,为了让不同蛋白质的PSSM具有可比性,研究人员通常会对初始的PSSM进行不同方式的标准化,标准化的方式也可以有不同的选择[16-17, 20]。

|
多个特征的组合一般可以提升预测的准确率,但同时也可能因为特征冗余带来噪声干扰,使模型过拟合并显著增加数据分析的存储需求和计算成本[31]。为了克服这些挑战,有必要进行特征筛选以获得最优特征子集。构建ECM蛋白质预测工具常用的特征筛选一般包括2个步骤:特征重要性打分(Feature importance score,FIS)和增量特征选择(Incremental feature selection,IFS)。
FIS是指通过特征打分算法对所有特征进行重要性打分并按照评分由高到低进行排序。现有ECM蛋白质预测工具中采用的算法包括:平均精度减少(Mean decrease accuracy,MDA)[14]、最大相关最小冗余(Maximum relevance minimum redundancy,MRMR)[15]、费希尔-马尔可夫选择器(Fisher-Markov selector)[16]、信息增益率(Information gain ratio,IGR)[17]和二元动物迁徙(Binary animal migration)[19]等。
基于FIS得到有序特征列表后,再通过IFS确定分类模型特征。该过程始于一个空特征子集,按照特征重要性的顺序由高到低依次加入,每加入一个特征,就生成一个新的特征子集(N个特征将生成N个特征子集)。预测表现最佳、同时包含更少特征的特征子集被认为是最优特征子集[17]。
1.4 学习分类经过特征提取和特征筛选两个步骤后,数据集中的每一个蛋白质序列都由一个最优特征向量来表征。基于最优特征向量和它们的分类标签(ECM或non-ECM),运用机器学习分类算法进行训练建模,最常用的算法是随机森林(Random forest,RF)[14-15, 17, 19, 32]和支持向量机(Support vector machine,SVM)[16, 20, 33]。随机森林能够同时处理连续型变量和离散型变量,并且处理速度较快,它对异常值和噪声有较好的容忍度,不易于过拟合,是一种很有优势的机器学习算法;但是当训练数据集不均衡时,随机森林会倾向于将样本预测为数目较多的类别[32]。支持向量机能更好地识别高维模式,同时在面对非线性和小样本问题时有更好的表现,它能够找到全局最优解,具有较优的泛化能力;但是支持向量机的分类预测效果比较依赖于核函数的选择,同时其运算效率也有待改进[34]。除此之外,Ali团队还尝试使用K近邻算法(K nearest neighbor,KNN)[35]作为ECM蛋白质预测工具的分类算法[18]。
在统计预测中,如果仅简单给出一个预测模型的成功率,而不说明使用的交叉验证方法,那么这样的结果是没有意义的[21]。常用的交叉验证方法包括:独立数据集检验(Independent dataset test)、K-fold交叉检验(K-fold cross validation)和Jackknife检验(Jackknife test)。相比于独立数据集检验和K-fold交叉检验,Jackknife检验的优势在于可以有效避免随意性的问题,即对于一个给定的原始数据集,Jackknife检验的结果是唯一的。但是Jackknife检验也有自身的劣势,当原始数据集中蛋白质序列的两两相似度超过25%时,Jackknife检验估计的成功率过高,同时Jackknife检验相对于其他两种交叉验证方式更耗时。现有ECM预测工具普遍采用的标准数据集中蛋白-蛋白序列的相似度都超过了25%的临界线,采用Jackknife检验会有高估成功率的风险且比较耗时,因此大部分预测工具都采用了独立数据集检验[15-16, 19]或K-fold交叉检验[14, 17-18]。
1.5 模型性能评估用于评估模型性能的参数一般包括敏感度(Sensitivity,Sn)、特异度(Specificity,Sp)、准确率(Accuracy,Acc)、Matthew相关系数(Matthew’s correlation coefficient,MCC)和受试者特征曲线(Receiver operating curve,ROC)。上述参数都可以通过真阳性(True positive,TP)、假阴性(False negative,FN)、真阴性(True negative,TN)和假阳性(False positive,FP) 4个指标来表示。
Sn指正确将阳性数据集中的样本预测为ECM蛋白质的比例:
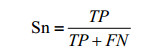
|
Sp指正确将阴性数据集中的样本预测为non-ECM蛋白质的比例:

|
Acc指正确预测数据集中ECM蛋白质和non-ECM蛋白质的比例:

|
在数据集不均衡的时候,MCC也是一个不错的评估模型性能的参数。它的取值范围是(−1, 1),MCC=1代表模型能正确地预测所有的样本,MCC=−1代表模型将所有的样本都预测错误,MCC=0代表模型在随机的进行预测。它的表示方式为:
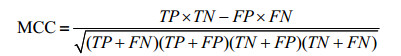
|
以上评估参数有一个共同的缺点:它们的取值依赖于阈值的选择,设置不同的阈值可能会出现不同的结果。而ROC则不受限于阈值,它是一条以Sn为纵轴,以(1−Sp)为横轴的曲线,通过计算曲线下面积(Area under the curve,AUC),可以有效衡量预测模型的性能。
2 ECM蛋白质预测工具的研究进展及存在的问题 2.1 ECM蛋白质预测工具的研究进展研究人员在研发ECM蛋白质预测工具的过程中,逐步发现并解决了很多问题,包括:探索数据集不均衡时的建模策略和评估方法,发掘能够显著预测ECM蛋白质的特征,对机器学习分类算法进行调参以获得更优的预测性能等等,这些研究成果对今后ECM蛋白质预测工具的发展和改进有重要的启示作用。目前已经发展的ECM蛋白质预测工具如表 1所示,本节将对这些工具的主要特点进行总结。
| Tool | Release date | Dataset | Extracted features | Feature selection method | Machine learning model | Cross-validation method |
| ECMPP | 2010 | Jung | SI/EI | Mean decrease accuracy | Random forest | 5-fold cross validation |
| ECMPred | 2013 | Kandaswamy | SI/PP | Maximum relevance Minimum redundancy | Random forest | Independent dataset test |
| PECM | 2014 | Kandaswamy | EI/SI/PP | Fisher-Markov selector | Support vector machine | Independent dataset test |
| IECMP | 2015 | Kandaswamy | SI/PP/EI/SI | Information gain ratio | Random forest | 10-fold cross validation |
| ECMP-HybKNN | 2016 | Kandaswamy | SI | Maximum relevance Minimum redundancy | K nearest neighbor | 10-fold cross validation |
| BAMORF | 2017 | Kandaswamy | SI/PP | Binary animal migration | Random forest | Independent dataset test |
| TargetECMP | 2018 | Kandaswamy | EI | -- | Support vector machine | Jackknife test |
| Jung: the standard dataset used by Jung et al to build ECMPP in 2010; Kandaswamy: the standard dataset used by Kandaswamy et al to build EcmPred in 2013; SI: extracted features based on the sequence information; EI: extracted features based on the evolutionary information; SI: extracted features based on the structural information; PP: extracted features based on physicochemical properties. | ||||||
ECMPP是较早通过机器学习建模对ECM蛋白质进行预测的工具[14],在ECM蛋白质预测研究领域具有开创性意义。在标准数据集构建方面,虽然后来的研究者广泛采用的是EcmPred工具发展的标准数据集,但是EcmPred数据集的构建思路和ECMPP完全一致:都是以Swiss-Prot中的多细胞动物分泌蛋白为初始数据集,认为有ECM注释是ECM蛋白质,没有ECM注释则为non-ECM蛋白质,去冗余之后形成最终的金标准数据集。因此可以认为ECMPP也是ECM蛋白质预测工具标准数据集构建思路的开创者。
EcmPred的最大贡献在于构建了一个被后来的ECM蛋白质预测工具研究者所广泛采用的标准数据集[15]。但是该数据集存在数据不均衡的问题,即阳性数据集的样本数目(445)远少于阴性数据集的样本数目(4 187)。如果采用不均衡的数据集进行模型训练,则会导致对小样本数据(即阳性数据)的预测精度变差[17]。针对这个问题,Kandaswamy等[15]提出了解决方案:从原始数据集中分别随机选取同等数目的ECM样本(300)和non-ECM样本(300)进入训练集,使得训练集中的阴性数据和阳性数据的数目相同。这种方法避免了模型在阳性数据集中预测精度较差的问题,却不能充分利用原始数据集的样本信息,只能作为一个初步的解决方案。
PECM首次使用了PSSM特征[16],并采用了SVM分类算法。相比于一般的特征,PSSM的批量提取一方面需要使用特定的工具(比如PSI-BLAST)和合适的背景库(比如SwissProt),另一方面需要耗费较多的时间(时间长短和背景库大小成正比)。但是实践证明这些代价是值得的,因为PSSM确实是预测ECM蛋白质的一个重要特征,在随后发展的ECM蛋白质预测工具研究中被广泛使用。SVM的一个特点是:C和γ这两个参数对模型影响很大,因此调参可以显著改善模型的性能。研究者普遍采用网格搜索策略(Grid search strategy)进行调参[16, 20]。
IECMP引入均衡准确率(Balanced accuracy,BAcc)来衡量预测模型的性能[17],同时使用集成分类器应对数据集不均衡问题。在均衡数据集中,Acc能较好地反映模型的总体性能。但是由于标准数据集中non-ECM蛋白质的数目远超过ECM蛋白质,模型会更倾向于将某个样本判定为non-ECM,导致模型在Acc很高的前提下,Sn依旧很低,使得Acc不能很好地反映模型的总体性能,因此在不均衡的数据集中,BAcc比Acc能更好地反映模型的总体性能。

|
为了解决数据集不均衡问题,同时能够充分利用原始数据的样本信息,IECMP提出了集成分类器的思路:将训练集中的阴性数据集随机均分为11个阴性数据子集,使得每个阴性数据子集的数目和训练集中的阳性数据接近。用这11个阴性数据子集分别和阳性数据集作为训练集来构建11个ECM蛋白质预测模型,然后应用这11个预测模型分别对测试集中的样本进行预测并投票,采用多数投票结果作为该测试样本的最终预测结果。实验结果表明使用集成分类器的预测表现要优于不使用集成分类器。
ECMP-HybKNN利用易提取的特征构建了一个高效的预测工具[18]。它仅选择了二肽组成(DPC)和伪氨基酸组成(PseAAC)作为提取的特征,这两个特征提取较为简单,因而计算效率也相应提高。
TargetECMP首次通过仅采用一个分类特征来构建ECM蛋白质预测工具[20],也取得了比较好的预测结果。以往的ECM蛋白质预测工具往往整合多个特征以反映更多的区分信息,而TargetECMP仅采用了基于灰色系统模型(Grey system model)[36-37]提取的进化信息(GreyPSSM),就得到了相当可观的模型性能。可能存在以下原因:Ⅰ) GreyPSSM确实是一个比较出色的ECM蛋白质预测特征;Ⅱ) TargetECMP通过对SVM分类器进行调参改善了模型性能。
2.2 ECM蛋白质预测工具现存的问题 2.2.1 标准数据集的更新问题截至目前,构建ECM蛋白质预测工具所使用的标准数据集依旧是Kandaswamy等于2013年提取的数据集[15]。该数据集在ECM蛋白质预测工具的构建中发挥了不可替代的作用,对推动ECM蛋白质预测工具的发展作出了巨大的贡献,同时该数据集也有一些值得改进之处。
第一,判定ECM和非ECM的标准。该数据集选取了SwissProt数据库中有ECM注释的分泌蛋白作为ECM,没有ECM注释的分泌蛋白为non-ECM。但是,一些分泌蛋白虽然没有注释为ECM,可能只是尚未得到实验研究,它本身由于可以被分泌到细胞外,理论上有可能为ECM或者ECM相关的蛋白质,因此目前判定non-ECM的标准很可能会将一些实际为ECM的蛋白判定为non-ECM,笔者认为从非分泌蛋白中挑选non-ECM可能是更为合理的选择。
第二,标准数据集中多物种混杂。该标准数据集从多细胞动物的分泌蛋白挑选而来,包含了多个物种的序列,个别物种仅含有少数几个蛋白质。多物种数据集使得训练得到的模型有更好的泛化能力,但是在预测某个特定物种蛋白质时,仅以该物种数据训练的模型将比多物种模型有更好的特异性,预测能力更强。因此笔者建议针对不同物种分别建立标准数据集,构建不同物种的ECM蛋白质预测工具。
2.2.2 特征提取的问题现有的ECM蛋白质预测工具大都选择了基于蛋白质序列信息、进化信息和理化性质计算得到的特征,但其中大部分不是ECM蛋白质功能特异相关的特征,这些特征可以用于预测ECM蛋白质,同样也能用于预测其他的蛋白质属性。这些非特异性特征能够反映ECM蛋白质足够多的信息,但是如若能合理利用ECM蛋白质特异的特征,将对提升ECM蛋白质预测性能有更大的帮助。
另一方面,大量研究指出ECM蛋白质普遍拥有一些保守的特征结构域,同时含有这些结构域的蛋白质有极大概率为ECM蛋白质[3]。Naba等曾对这些特征结构域做过总结[9],但是目前尚未有预测工具利用过此特征结构域列表。笔者认为将来的ECM蛋白质预测工具应将此作为重要的特征进行考虑。
2.2.3 分类算法的问题已有ECM蛋白质预测工具全部基于传统的机器学习进行分类预测,而方兴未艾的深度学习尚未应用到该领域[38]。机器学习算法的准确性很大程度上依赖于良好的特征提取,这个过程通过人工完成,因此提取一组好的特征需要相关研究者对问题有相当深入的认知,这就需要花费巨大的时间和人力成本;而深度学习算法的一大特征便是能够自动学习有用的特征,因此将大大节省时间和人力成本,同时有可能得到更好的预测准确性。
制约深度学习算法在ECM蛋白质预测领域应用的一个重要原因可能是深度学习需要较大的数据集,而目前ECM蛋白质预测领域的金标准数据集规模仍较小。但是,一方面现有标准数据集可以不断完善,在提升数据集准确度的同时扩大规模;另一方面,深度学习已有在小样本上应用成功的案例[39]。因此,将来ECM蛋白质预测工具的发展完全有可能也有必要引进深度学习算法,以期实现对ECM蛋白质预测的更大突破。
2.2.4 工具的可用性问题在已发展的ECM蛋白质预测模型中,IECMP及其之前的工作都将预测模型做成了线上工具,但是目前大多已无法使用;IECMP之后的预测模型则没有再提供用户可以直接使用的工具。因此,开发用户体验良好的ECM蛋白质预测工具并进行长期稳定的维护,是一项比较重要的工作,它不仅可以方便生物学背景的研究人员对相关蛋白直接进行预测,还可以方便后续的ECM蛋白质预测工具开发者与已发展的预测工具进行比较。因此,将ECM蛋白质预测模型开发为好用的工具应是相关研究人员要达到的基本目标;在此基础上应对开发的可用工具进行持续的维护,以保证在相当长一段时间内,用户在需要时可以调取使用;当然,进一步将该工具开发为用户友好型的线上工具则更好,这将为相关用户和研究者的使用带来极大便利。
3 总结构建ECM蛋白质预测工具对于ECM蛋白质的研究具有重要意义。基于“选取数据集——特征提取——特征筛选——学习分类——模型性能评估”这个基本流程,研究者已经开发了一系列ECM蛋白质预测工具。前人的研究启示我们,通过集成分类器建模和通过均衡准确率进行评估可以较好地应对数据集不均衡问题;二肽组成、伪氨基酸组成、理化性质和位置特异性得分矩阵等特征在ECM蛋白质预测中起到重要作用;特征筛选可以对特征去冗余,从而提升模型性能;对机器学习分类算法进行调参可以获得更优的预测性能。
目前的ECM蛋白质预测工具面临4个主要问题:首先,标准数据集存在分类标准不严格和物种混合的问题,有必要对数据集进行更新;其次,已有工具提取的特征缺少ECM蛋白质特异性特征,笔者认为有必要引进基于实验的ECM蛋白质特异性特征以提高预测性能;再次,可以尝试引进深度学习算法来优化ECM蛋白质的预测建模;最后,目前ECM蛋白质预测领域普遍缺乏便捷的线上工具,对ECM蛋白质预测产生了不利影响,该领域研究者有必要配套开发ECM蛋白质预测的线上工具。
| [1] |
Naba A, Clauser KR, Ding HM, et al. The extracellular matrix: tools and insights for the "omics" era. Matrix Biol, 2016, 49: 10-24. DOI:10.1016/j.matbio.2015.06.003 |
| [2] |
Hynes RO, Naba A. Overview of the matrisome—an inventory of extracellular matrix constituents and functions. Cold Spring Harb Perspect Biol, 2012, 4(1): a004903. |
| [3] |
Hynes RO. The extracellular matrix: not just pretty fibrils. Science, 2009, 326(5957): 1216-1219. DOI:10.1126/science.1176009 |
| [4] |
Frangogiannis NG. The extracellular matrix in myocardial injury, repair, and remodeling. J Clin Invest, 2017, 127(5): 1600-1612. DOI:10.1172/JCI87491 |
| [5] |
Tokhmafshan F, Brophy PD, Gbadegesin RA, et al. Vesicoureteral reflux and the extracellular matrix connection. Pediatr Nephrol, 2017, 32(4): 565-576. DOI:10.1007/s00467-016-3386-5 |
| [6] |
Lim J, Grafe I, Alexander S, et al. Genetic causes and mechanisms of Osteogenesis Imperfecta. Bone, 2017, 102: 40-49. DOI:10.1016/j.bone.2017.02.004 |
| [7] |
Bateman JF, Boot-Handford RP, Lamandé SR. Genetic diseases of connective tissues: cellular and extracellular effects of ECM mutations. Nat Rev Genet, 2009, 10(3): 173. DOI:10.1038/nrg2520 |
| [8] |
Walker C, Mojares E, Del Rio Hernandez A. Role of extracellular matrix in development and cancer progression. Int J Mol Sci, 2018, 19(10): 3028. DOI:10.3390/ijms19103028 |
| [9] |
Naba A, Clauser KR, Hoersch S, et al. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol Cell Proteomics, 2012, 11(4): M111.014647. DOI:10.1074/mcp.M111.014647 |
| [10] |
Lennon R, Byron A, Humphries JD, et al. Global analysis reveals the complexity of the human glomerular extracellular matrix. J Am Soc Nephrol, 2014, 25(5): 939-951. DOI:10.1681/ASN.2013030233 |
| [11] |
Mayorca-Guiliani AE, Madsen CD, Cox TR, et al. ISDoT: in situ decellularization of tissues for high-resolution imaging and proteomic analysis of native extracellular matrix. Nat Med, 2017, 23(7): 890-898. DOI:10.1038/nm.4352 |
| [12] |
Gopal S, Veracini L, Grall D, et al. Fibronectin-guided migration of carcinoma collectives. Nat Commun, 2017, 8: 14105. DOI:10.1038/ncomms14105 |
| [13] |
Åhrman E, Hallgren O, Malmström L, et al. Quantitative proteomic characterization of the lung extracellular matrix in chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. J Proteomics, 2018, 189: 23-33. DOI:10.1016/j.jprot.2018.02.027 |
| [14] |
Jung J, Ryu T, Hwang Y, et al. Prediction of extracellular matrix proteins based on distinctive sequence and domain characteristics. J Comput Biol, 2010, 17(1): 97-105. DOI:10.1089/cmb.2008.0236 |
| [15] |
Kandaswamy KK, Pugalenthi G, Kalies KU, et al. EcmPred: prediction of extracellular matrix proteins based on random forest with maximum relevance minimum redundancy feature selection. J Theor Biol, 2013, 317: 377-383. DOI:10.1016/j.jtbi.2012.10.015 |
| [16] |
Zhang J, Sun PP, Zhao XW, et al. PECM: prediction of extracellular matrix proteins using the concept of Chou's pseudo amino acid composition. J Theor Biol, 2014, 363: 412-418. DOI:10.1016/j.jtbi.2014.08.002 |
| [17] |
Yang RT, Zhang CJ, Gao R, et al. An ensemble method with hybrid features to identify extracellular matrix proteins. PLoS ONE, 2015, 10(2): e0117804. DOI:10.1371/journal.pone.0117804 |
| [18] |
Ali F, Hayat M. Machine learning approaches for discrimination of Extracellular Matrix proteins using hybrid feature space. J Theor Biol, 2016, 403: 30-37. DOI:10.1016/j.jtbi.2016.05.011 |
| [19] |
Guan LZ, Zhang SW, Xu HQ. BAMORF: a Novel computational method for predicting the extracellular matrix proteins. IEEE Access, 2017, 5: 18498-18505. DOI:10.1109/ACCESS.2017.2751499 |
| [20] |
Kabir M, Ahmad S, Iqbal M, et al. Improving prediction of extracellular matrix proteins using evolutionary information via a grey system model and asymmetric under-sampling technique. Chemom Intell Lab Syst, 2018, 174: 22-32. DOI:10.1016/j.chemolab.2018.01.004 |
| [21] |
Chou KC. Some remarks on protein attribute prediction and pseudo amino acid composition. J Theor Biol, 2011, 273(1): 236-247. DOI:10.1016/j.jtbi.2010.12.024 |
| [22] |
Wang L, Zhao Y, Chen YH, et al. The effect of three novel feature extraction methods on the prediction of the subcellular localization of multi-site virus proteins. Bioengineered, 2018, 9(1): 196-202. DOI:10.1080/21655979.2017.1373536 |
| [23] |
Hayat M, Tahir M, Khan SA. Prediction of protein structure classes using hybrid space of multi-profile Bayes and bi-gram probability feature spaces. J Theor Biol, 2014, 346: 8-15. DOI:10.1016/j.jtbi.2013.12.015 |
| [24] |
Ahmad J, Hayat M. MFSC: multi-voting based feature selection for classification of Golgi proteins by adopting the general form of Chou's PseAAC components. J Theor Biol, 2019, 463: 99-109. DOI:10.1016/j.jtbi.2018.12.017 |
| [25] |
Chou KC. Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins, 2001, 43(3): 246-255. |
| [26] |
Kawashima S, Pokarowski P, Pokarowska M, et al. AAindex: amino acid index database, progress report 2008. Nucleic Acids Res, 2008, 36(Database issue): D202-D205. |
| [27] |
Chou KC, Shen HB. Large-scale plant protein subcellular location prediction. J Cell Biochem, 2007, 100(3): 665-678. DOI:10.1002/(ISSN)1097-4644 |
| [28] |
Zuo YC, Peng Y, Liu L, et al. Predicting peroxidase subcellular location by hybridizing different descriptors of Chou' pseudo amino acid patterns. Anal Biochem, 2014, 458: 14-19. DOI:10.1016/j.ab.2014.04.032 |
| [29] |
Ding SY, Yan SJ, Qi SH, et al. A protein structural classes prediction method based on PSI-BLAST profile. J Theor Biol, 2014, 353: 19-23. DOI:10.1016/j.jtbi.2014.02.034 |
| [30] |
Altschul SF, Madden TL, Sch ffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res, 1997, 25(17): 3389-3402. DOI:10.1093/nar/25.17.3389 |
| [31] |
Li JD, Cheng KW, Wang SH, et al. Feature selection: a data perspective. ACM Comput Surv, 2018, 50(6): 94. |
| [32] |
Breiman L. Random forests. Mach Learn, 2001, 45(1): 5-32. DOI:10.1023/A:1010933404324 |
| [33] |
Vapnik V. Statistical Learning Theory. New York: John Wiley & Sons, 1998.
|
| [34] |
Burges CJC. A tutorial on support vector machines for pattern recognition. Data Min Knowl Discov, 1998, 2(2): 121-167. DOI:10.1023/A:1009715923555 |
| [35] |
Akkus A, Güvenir HA. K nearest neighbor classification on feature projections//Proceedings of the Thirteenth International Conference on International Conference on Machine Learning. Bari, Italy: ACM, 1996.
|
| [36] |
Matsuda S, Vert JP, Saigo H, et al. A novel representation of protein sequences for prediction of subcellular location using support vector machines. Protein Sci, 2005, 14(11): 2804-2813. DOI:10.1110/ps.051597405 |
| [37] |
Lin WZ, Fang JA, Xiao X, et al. iLoc-Animal: a multi-label learning classifier for predicting subcellular localization of animal proteins. Mol Biosyst, 2013, 9(4): 634-644. DOI:10.1039/c3mb25466f |
| [38] |
LeCun Y, Bengio Y, Hinton G. Deep learning. Nature, 2015, 521(7553): 436-444. DOI:10.1038/nature14539 |
| [39] |
Ng HW, Nguyen VD, Vonikakis V, et al. Deep learning for emotion recognition on small datasets using transfer learning//Proceedings of the 2015 ACM on International Conference on Multimodal Interaction. Seattle, Washington, USA: ACM, 2015.
|