 2018, Vol. 34
2018, Vol. 34中国科学院微生物研究所、中国微生物学会主办
文章信息
- 周茂志, 唐存多, 许建和, 郁惠蕾
- Zhou Maozhi, Tang Cunduo, Xu Jianhe, Yu Huilei
- 新型扁桃酸消旋酶的基因挖掘和性能表征
- Genome mining and characterization of a new mandelate racemase
- 生物工程学报, 2018, 34(6): 897-905
- Chinese Journal of Biotechnology, 2018, 34(6): 897-905
- 10.13345/j.cjb.170441
-
文章历史
- Received: December 12, 2017
- Accepted: December 22, 2017

引用本文

|



2 南阳师范学院 昆虫生物反应器河南省工程实验室,河南 南阳 473061
2 Henan Provincial Engineering Laboratory of Insect Bio-reactor, Nanyang Normal University, Nanyang 473061, Henan, China
光学纯化合物可广泛应用于医药、香精香料和特殊化学品等制造行业, 其合成需求日益增加。在现代有机化学制造过程中, 部分单一构型化学品的合成仍然具有挑战性[1]。近年来有机催化、金属催化与生物催化技术的进步推动了光学纯化合物的制备, 其中外消旋体的催化动力学拆分仍然是工业制备手性化学品的主要方式之一[2]。然而, 此类传统的动力学拆分方法存在明显的缺陷, 即单个对映体的理论得率最高为50%。研究人员从过程经济性和绿色环保的角度考虑, 提出了一种理论得率可达到100%的拆分途径, 即动态动力学拆分或称作去消旋化。动态动力学拆分结合传统动力学拆分和原位消旋化两种方法将外消旋形式的底物完全转化成单一构型的目标产物, 理论得率可达到100%, 同时省去反应中间体的分离步骤[3-5]。相比于化学法原位消旋化过程, 酶促消旋化反应的条件更加温和, 往往在常温常压及中性pH环境下完成, 可以避免缩合、重排等副反应发生, 在工业应用中可能更具优势[6-7]。消旋酶(Racemase)是催化互为对映异构体的两种化合物之间相互转化的酶类, 属于异构酶家族, 酶学分类号为EC 5.1.X.X。尽管目前所报道的消旋酶种类非常有限, 但其在合成中的应用前景不可忽视。
扁桃酸消旋酶(EC 5.1.2.2)可催化扁桃酸对映异构体之间的相互转化[8-11]。2012年奥地利生物有机化学家Faber K.教授报道了由扁桃酸脱氢酶、氨基酸脱氢酶与扁桃酸消旋酶级联催化的一锅煮反应, 从外消旋扁桃酸出发直接制备获得光学纯L-苯甘氨酸, 最终产物得率高达94%, ee值大于97%[12]。该级联反应条件温和、选择性高, 成为L-苯甘氨酸及其衍生物合成的一条新途径[13]。所得产物可用于合成β-内酰胺类抗生素及抗血小板类药物, 在医药工业有着重要价值。因此进一步挖掘和研究新的扁桃酸消旋酶资源, 对于拓宽上述多酶级联反应的催化效率和应用范围具有重要意义。文中以文献报道的唯一扁桃酸消旋酶序列[4]为探针, 通过基因数据挖掘方法寻找新型的扁桃酸消旋酶, 并研究其酶学性质和催化动力学。
1 材料与方法 1.1 材料 1.1.1 主要试剂与仪器扁桃酸(Mandelic acid, MA)与邻氯扁桃酸(2-chloromandelic acid, 2-Cl MA)由江西科苑生物药业有限公司提供, 分析纯; 间氯扁桃酸与对氯扁桃酸由本实验室制备, 纯度 > 97%;异丙醇购自迪马科技有限公司, 色谱纯。如非特殊说明, 实验用其他试剂和材料均来源于市售, 试剂纯度均为分析纯; 消旋酶催化反应的进度通过岛津LC-2100A高效液相色谱分析检测; 蛋白浓度通过BCA试剂盒与BioTek Power Wave XS2酶标仪结合测定; 核酸浓度定量使用ThermoFisher紫外分光光度计NanoDrop ND-2000测定。
1.1.2 菌株与质粒所用的菌种和质粒由本实验室保藏。
1.1.3 培养基LB培养基:酵母提取物5 g/L, 蛋白胨10 g/L, 氯化钠10 g/L; 调节pH至7.0, 在121 ℃灭菌20 min。在液体LB培养基中加入1.5%-2%的琼脂配成固体培养基, 相同条件灭菌使用。
1.2 方法 1.2.1 扁桃酸消旋酶的基因挖掘与筛选本文以扁桃酸为目标底物, 寻找对其有活力的消旋酶。文献调研发现, 已经报道的对此类底物有消旋活力的只有来源于恶臭假单胞菌Pseudomonas putida ATCC 12633扁桃酸消旋酶PpMR (Uniprot登录号P11444)。本研究以该酶作为探针, 将其蛋白序列在NCBI数据库中进行BLASTp。为保证序列多样性, 挑选与探针序列相似性在20%-60%左右的候选序列作为待克隆蛋白, 这些序列功能上并没有经过详细注释和验证。对虚拟筛选得到的序列分别进行克隆和诱导表达。
考察粗酶催化底物转化的情况, 1 mL反应体系(2 mL圆底离心管)如下:1 mmol/L单一构型的扁桃酸或邻氯扁桃酸, 100 μL上清酶液, 3.3 mmol/L MgCl2·6H2O, 840 μL Tris-HCl缓冲液(pH 7.5, 100 mmol/L)。混匀之后将2 mL离心管置于振荡器上, 30 ℃、1 000 r/min反应12 h, 加入适量2 mol/L硫酸终止反应, 然后再用等体积的乙酸乙酯萃取处理后的反应液, 14 000 r/min离心2 min吸取上层有机相液体, 加入少许无水硫酸钠干燥8 h以上, 干燥结束后样品经离心过滤处理, 通过液相色谱法检测底物转化情况。综合比较目标蛋白的催化效率和表达情况, 选择最优的消旋酶进行进一步实验。
1.2.2 重组消旋酶的纯化本实验选择pET28a作为原核表达载体, 该质粒在表达目的蛋白过程中可在蛋白C端或N端引入多聚组氨酸纯化标签, 基于亲和层析原理利用HisTrap预装柱(GE Healthcare)可直接一步捕获重组菌体破碎上清的目标蛋白。因为该酶在活性中心含有金属离子, 纯化时选择Tris-HCl缓冲体系。纯化过程主要由4个步骤组成:漂洗、平衡、上样和洗脱; 洗脱收集的样品经过SDS-PAGE分析选择电泳纯度较高的样品进行透析换液, 获得的纯酶低温储存用于该酶的性质表征。
1.2.3 分析方法及扁桃酸消旋酶纯酶活力的测定扁桃酸消旋酶纯酶活力的测定是通过反应检测产物的生成速率计算其活力。1 mL反应体系应包括100 mmol/L的Tris-HCl缓冲液(pH 7.5)、2 mmol/L底物、3.3 mmol/L镁离子、适量纯酶。30 ℃、1 000 r/min离心, 反应特定的时间, 终止反应分析产物的生成量, 计算其比活力。
采用液相色谱法分析反应过程中底物的转化和产物的生成量。所用检测条件:色谱柱为CHIRALCEL®OJ-H (4.6 mm×250 mm), 检测波长228 nm, 柱温30 ℃; 流动相为正己烷/异丙醇(92:8, V/V), 添加1‰三氟乙酸; 流速为1 mL/min。(R)-扁桃酸出峰时间为21.5 min, (S)-扁桃酸出峰时间为24.9 min, (R)-邻氯扁桃酸出峰时间为21.4 min, (S)-邻氯扁桃酸出峰时间为26.4 min。
1.2.4 扁桃酸消旋酶最适温度及温度稳定性测定不同温度下该酶的催化效率, 反应体系如下(1 mL):2 mmol/L (R)-扁桃酸、3.3 mmol/L MgCl2·6H2O和适量纯酶, HEPES缓冲体系; 在1 000 r/min、不同温度(20、30、40、50、60 ℃)下反应15 min后, 加硫酸酸化终止反应, 等体积乙酸乙酯萃取, 离心后吸取上层置于通风厨中挥干, 加入异丙醇溶解并干燥处理样品, 液相分析反应结果, 从而计算不同温度下该酶的催化活力。
将纯酶液用HEPES缓冲液稀释成合适的浓度, 在30 ℃、40 ℃、50 ℃下分别保温, 不同的时间点取样, 按1.2.3所述方法进行残余活力的测定。研究其相对活力与时间的进程关系, 以未保温前的活力为基准点100%。对相对活力取对数与时间关系进行拟合, 计算不同温度下扁桃酸消旋酶催化活性的半衰期。
1.2.5 扁桃酸消旋酶最适pH的研究配置不同pH的缓冲液, 包括pH 4.0-6.0柠檬酸-柠檬酸钠缓冲液、pH 6.0-7.0磷酸盐缓冲液、pH 7.5-9.0 Tris-HCl缓冲液、pH 9.0-11.0甘氨酸-氢氧化钠缓冲液等。考察不同pH环境中该酶的催化活力, 按如下反应体系(1 mL):2 mmol/L (R)-扁桃酸、3.3 mmol/L MgCl2·6H2O和适量纯酶, 不同pH缓冲液补足至1 mL, 置于30 ℃、1 000 r/min反应15 min后加2 mol/L硫酸终止反应, 按照1.2.3所述方法, 分析该酶在不同pH缓冲条件中的活力。
1.2.6 添加剂对扁桃酸消旋酶活力的影响反应体系中加入数种不同的添加剂进行该酶的催化反应, 体系内组分如下:2 mmol/L (R)-扁桃酸、适量纯酶, 添加剂1 mmol/L, Tris-HCl缓冲体系; 30 ℃反应10 min, 终止反应, 处理和分析样品; 计算不同添加剂环境下的催化活力, 考察添加剂对该酶活力的影响。
1.2.7 扁桃酸消旋酶对其他底物的活力考察该酶对不同底物的活性, 反应底物浓度同为2 mmol/L, 测活及分析方法参见1.2.3。
1.2.8 扁桃酸消旋酶催化动力学研究对4种不同的底物[(R)-扁桃酸、(S)-扁桃酸、(S)-邻氯扁桃酸、(R)-邻氯扁桃酸]进行动力学的研究, 按照酶动力学研究标准方法, 设定不同的底物浓度值, 测定反应速率和时间的关系。采用Origin拟合动力学曲线, 得到所对应的催化动力学常数, 包括表征酶与底物亲和力的米氏常数Km与最大反应速率Vmax, 从而计算出酶针对不同分子的催化转换速率常数kcat值。
2 结果与分析 2.1 扁桃酸消旋酶的克隆表达与功能筛选目前唯一报道的扁桃酸消旋酶是来源于恶臭假单胞菌的PpMR, 经数十年的研究其分子结构和催化机理已经得到充分的解析[11-15]。PpMR催化扁桃酸消旋反应的转换频率(kcat值)约为500 s-1, 其米氏常数(Km值)约为1 mmol/L, 是目前研究高pKa值羟基羧酸酶促去质子化作用的经典催化剂范本[16-18]。
通过基因挖掘的手段[19], 成功克隆得到28个目标蛋白(表 1)。SDS-PAGE分析所表达的蛋白大小都在40 kDa左右, 与数据库预测的大小一致。对目标蛋白的功能筛选发现其中存在9个蛋白(Bj2、Ar、Eb2、Tf2、Ar2、Eb1、St3、Bj7、Cl)可催化扁桃酸的消旋反应, 但当以单一构型的邻氯扁桃酸为底物时, 只有两个酶蛋白(Ar、Bj2)可检测到催化活性, 但Bj2只转化很少量的底物, 而且表达量低、可溶性很差。与探针PpMR酶的综合性能相比, 来源于Agrobacterium radiobacter的扁桃酸消旋酶ArMR具有更好的可溶性表达和催化活力(表 2), 因此我们选择ArMR酶作为进步一研究的对象。其他8个扁桃酸消旋酶也增加了该类酶资源的有用文献信息, 为今后深入考察此类扁桃酸消旋酶打下了一定基础。
| Target proteins | Accession No. | Organism | Mass (kDa) | Identity to PpMR (%) |
| Bj2 | A0A038GMM5 | Burkholderia jiangsuensis | 39.1 | 57 |
| Ar | B9JQ28 | Agrobacterium radiobacter | 38.7 | 57 |
| Eb2 | D8MW22 | Erwinia billingiae | 48.0 | 27 |
| Tf2 | R9F6C1 | Thermobifida fusca | 38.7 | 33 |
| Ar2 | B9JAW7 | Agrobacterium radiobacter | 40.4 | 31 |
| Eb1 | D8MLK0 | Erwinia billingiae | 41.3 | 28 |
| St3 | Q96XF6 | Sulfolobus tokodaii | 42.2 | 23 |
| Bj7 | A0A038GY43 | Burkholderia jiangsuensis | 44.3 | 27 |
| Cl | D8GJQ9 | Clostridium ljungdahlii | 46.5 | 20 |
| At | W8FLW8 | Agrobacterium tumefaciens | 43.5 | 30 |
| Ph1 | G4RFL4 | Pelagibacterium halotolerans | 41.5 | 31 |
| Pc | C6DAG0 | Pectobacterium carotovorum | 43.8 | 30 |
| St1 | Q972M4 | Sulfolobus tokodaii | 42.2 | 29 |
| Ph2 | G4RFN1 | Pelagibacterium halotolerans | 41.5 | 30 |
| Pd | F4CW58 | Pseudonocardia dioxanivorans | 38.7 | 43 |
| Tf1 | Q47P67 | Thermobifida fusca | 38.7 | 32 |
| Lf | C0WZ23 | Lactobacillus fermentum | 46.0 | 20 |
| Ev | L0FZ31 | Echinicola vietnamensis | 50.4 | 23 |
| St2 | Q96XE5 | Sulfolobus tokodaii | 44.7 | 25 |
| St4 | Q96Y01 | Sulfolobus tokodaii | 39.4 | 26 |
| Bj1 | A0A038GLD1 | Burkholderia jiangsuensis | 40.3 | 30 |
| Bj3 | A0A038GS62 | Burkholderia jiangsuensis | 48.2 | 24 |
| Bj4 | A0A038GTM5 | Burkholderia jiangsuensis | 43.5 | 30 |
| Bj5 | A0A038GUG0 | Burkholderia jiangsuensis | 40.1 | 28 |
| Bj6 | A0A038GWG4 | Burkholderia jiangsuensis | 41.7 | 31 |
| Bj8 | A0A038H0N7 | Burkholderia jiangsuensis | 44.8 | 25 |
| Bj9 | A0A038H5T4 | Burkholderia jiangsuensis | 47.0 | 23 |
| Bj10 | A0A038H410 | Burkholderia jiangsuensis | 44.8 | 32 |
| The Proteins highlighted in bold type have showed the racemic activity. | ||||
| Enzyme[a] | Time (h) | Conversion (%) |
| ArMR | 2 | 1.8 |
| 12 | 12.5 | |
| PpMR | 2 | < 1[b] |
| 12 | 3.3 | |
| Reaction mixture (1 mL):CFE 100 μL, substrate 5 mmol/L, 30 ℃, shaken at 1 000 r/min.[a] Cell-free extract (CFE) was produced by sonic disruption of cell suspension (50 g/L, wet weight) in Tris-HCl buffer (pH 7.5, 100 mmol/L).[b] Undetectable using the assay procedure. | ||
基于亲和层析的原理, 通过Ni-NTA对ArMR进行纯化, 获得纯度较高的纯化酶用于该酶的酶学性质表征。如图 1所示, SDS-PAGE清楚地展示了该酶的表达与纯化效果, 可见大肠杆菌异源表达的ArMR酶部分可溶(泳道1、3), 但沉淀部分仍有可观量的包涵体蛋白有待进一步复性利用。纯化所得到的酶(泳道5)条带足够清晰单一, 符合酶学特性表征的要求。测定纯酶活力, 对(R)-扁桃酸的比活为320 U/mg。

|
| 图 1 扁桃酸消旋酶ArMR蛋白纯化SDS-PAGE分析 Figure 1 SDS-PAGE analysis of purified ArMR.lane M:low molecular weight protein marker; lanes 1 & 3:cell lysate supernatant of recombinant ArMR; lanes 2 & 4:cell lysate precipitate of recombinant ArMR; lane 5:the purified ArMR. |
| |
在本实验中, 研究最适温度和温度稳定性所选用的是pH 7.5的HEPES缓冲液, 因为Tris-HCl缓冲液的pH受温度影响较大。温度对ArMR酶活力的影响如图 2所示。随着温度的上升, 该酶活力呈现先上升后下降的趋势, 最适宜温度是50 ℃。在30 ℃和40 ℃时, 相对活力约为50%和70%, 但当温度降到20 ℃时只有20%左右的相对活力。在高温如60 ℃情况下, 仍然能有70%的相对活力。

|
| 图 2 温度对酶ArMR活力的影响 Figure 2 Effects of temperature on the activity of purified ArMR. |
| |
不同温度下热稳定性考察结果表明, ArMR纯酶在30 ℃、40 ℃与50 ℃下的半衰期分别为70.7、27.2和0.17 h。最适温度研究表明该酶在50 ℃时催化反应的效果最佳, 可其半衰期仅为12 min, 可能会限制未来该酶的实际应用, 因此ArMR的热稳定性还存着较大的提升空间。
2.3.2 ArMR催化消旋化的最适pH如图 3所示, pH对该酶的活力有明显影响, 整体趋势呈现钟罩状。在pH 6以下的酸性环境中基本没有消旋活力, 因为该酶催化机理符合双碱机制, 由典型的两个碱性氨基酸残基催化质子的得失[14], 推测较酸性的环境不利于质子转移, 明显降低催化活性。在碱性pH范围内该酶活力有较好的接纳范围, 甚至在pH为10的甘氨酸-氢氧化钠缓冲液中仍然保留着10%以上的相对活力。ArMR在pH 8左右的Tris-HCl缓冲液中表现出最大的活力。

|
| 图 3 pH对酶ArMR活力的影响 Figure 3 Effects of pH on the activity of purified ArMR. |
| |
考察了11种不同添加剂, 主要是考察二价金属离子对ArMR催化活力的影响。添加剂浓度设定为1 mmol/L, 结果如表 3所示。除了Zn2+对该酶有完全的失活作用, 其他添加剂的影响较小。二价镁离子和锰离子对酶有部分的激活作用, 其他金属离子包括二价铁离子、铜离子和钴离子等对酶活基本无影响。在体系中添加1 mmol/L的EDTA进行保温, 结果发现仍保留86%的相对活力。
| Additive (1 mmol/L) | Relative activity (%) |
| None | 100.0 |
| EDTA | 86.9±0.2 |
| Na+ | 96.2±0.1 |
| K+ | 96.9±0.3 |
| Mg2+ | 121.3±0.6 |
| Cu2+ | 91.3±0.1 |
| Ca2+ | 97.5±0.0 |
| Ba2+ | 95.6±0.2 |
| Fe2+ | 83.8±0.3 |
| Co2+ | 97.5±0.1 |
| Mn2+ | 114.0±1.0 |
| Zn2+ | 0.0 |
测定ArMR纯酶对不同底物的相对活力结果, 比较其对不同底物的活力与PpMR的差异。结果如表 4所示, 可见这两个扁桃酸消旋酶在底物的偏好性上有明显的差异。PpMR对对氯扁桃酸活力要好于间氯扁桃酸[20]; 相比于扁桃酸, 该酶对间氯扁桃酸的相对活力有所下降。而ArMR对间氯和对氯底物的活力都比扁桃酸要好, 同时对间氯底物的活性要高于对氯扁桃酸。两者的共同之处在于它们对邻氯扁桃酸的消旋化活力仍然严重偏低, 不及扁桃酸的千分之一, 未来需要通过定点突变[21]等技术加以改造。由于这两个酶的蛋白序列一致性只有57%, 因此在催化的底物专一性上出现一定差异也就不难理解。
| Substrates | Relative activity (%) | |
| ArMR | PpMR | |
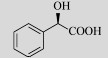
|
100 | 100 |
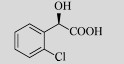
|
< 0.1 | < 0.1 |
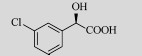
|
287 | 61 |

|
158 | 326 |
选择4种不同的底物进行动力学考察, 即(R)-扁桃酸、(S)-扁桃酸、(R)-邻氯扁桃酸和(S)-邻氯扁桃酸。反应体系如下:不同浓度的该底物, 3.3 mmol/L镁离子, 添加适量的ArMR纯酶, 缓冲液浓度为Tris-HCl缓冲液(100 mmol/L, pH 7.8), 在30 ℃、1 000 r/min振荡器上反应若干时间, 测定该酶催化不同浓度底物的反应初速度。利用Origin软件对不同底物浓度的反应初速度进行非线性拟合, 求得Km和Vmax值, 根据所添加的纯酶浓度计算酶的转化频率(kcat值), 如表 5所示。
| Substrate | Direction | Km (mmol/L) | Vmax (μmol/(min·mg)) | kcat (s-1) | kcat/Km (L/(mmol·s)) |
| MA | R→S | 1.44±0.19 | 582.00±27.00 | 410.00 | 285.000 |
| S→R | 0.81±0.07 | 310.00±8.00 | 218.00 | 270.000 | |
| 2-Cl MA | R→S | 6.48±0.88 | 0.31±0.02 | 0.22 | 0.034 |
| S→R | 6.37±0.79 | 0.33±0.02 | 0.23 | 0.036 |
表 5列出ArMR催化4种不同底物的动力学参数, ArMR对扁桃酸的Km值在1 mmol/L左右, 对R型底物的kcat为410 s-1, 对S型底物的kcat为218 s-1。催化R/S两种不同构型扁桃酸的最大反应速度分别为582、310 μmol/(min·mg)。同时该酶对邻氯扁桃酸的Km值约为6 mmol/L, 对R型邻氯扁桃酸的kcat值为0.22 s-1, 对S型邻氯扁桃酸的kcat为0.23 s-1。催化R/S两种不同构型邻氯扁桃酸的最大反应速度分别为0.31、0.33 μmol/(min·mg), kcat/Km值分别为0.034 L/(mmol·s)和0.036 L/(mmol·s)。而PpMR纯酶催化两种不同构型邻氯扁桃酸的kcat/Km分别为0.060 L/(mmol·s)和0.056 L/(mmol·s), PpMR纯酶的催化效率稍稍高于ArMR[22]。
3 讨论计算机信息技术的进步使生物科学同样步入了大数据时代, 这大大加速了生物信息学的发展, 基因和蛋白数据库容量得到飞速提高。如何在浩瀚的生物信息海洋中快速定位获得所需要的信息, 是目前生物资源更充分利用所遇到的难题。针对类似问题, 新的数据挖掘技术应运而生。基因组挖掘(Genome mining)利用已知的探针在基因和蛋白质数据库中进行序列比对和分析, 能够快速挖掘到研究所需的目的序列。NCBI数据库中现已存储数万条完整的微生物基因组序列, 其中含有大量未被发现的新酶。在生物信息学工具的辅助下, 如多序列比对与同源序列查找等, 本研究利用文献报道的来自于恶臭假单胞菌Pseudomonas putida ATCC 12633中的扁桃酸消旋酶PpMR为探针, 在数据库中进行目标序列的虚拟筛选, 利用分子生物学手段进行异源克隆表达, 以目标底物扁桃酸和邻氯扁桃酸进行功能筛选, 成功克隆得到9个新型的扁桃酸消旋酶, 丰富了α-羟基酸类手性化合物消旋酶的品种数目, 为未来深入研究此类手性合成工具酶提供了便利和参考。针对底物扁桃酸和邻氯扁桃酸, 对催化性能较好的消旋酶ArMR进行了表征, 并测定了动力学和热力学等理化特征参数。结合新型的酶促外消旋化和传统合成技术的去消旋化过程, 可望满足化学工业发展的绿色化需求, 而该酶对非天然底物的催化活力, 显示其可用于拓展合成L-苯甘氨酸及其衍生物的酶促级联反应, 有望发展成为合成重要非天然氨基酸类化合物的新途径。
| [1] | Rachwalski M, Vermue N, Rutjes FPJT. Recent advances in enzymatic and chemical deracemisation of racemic compounds. Chem Soc Rev, 2013, 42(24): 9268–9282. DOI: 10.1039/c3cs60175g |
| [2] | Lin GQ, You QD, Cheng JF. Chiral Drugs:Chemistry and Biological Action. New York: John Wiley & Sons, 2011: 3-28. |
| [3] | Turner NJ. Deracemisation methods. Curr Opin Chem Biol, 2010, 14(2): 115–121. DOI: 10.1016/j.cbpa.2009.11.027 |
| [4] | Caddick S, Jenkins K. Dynamic resolutions in asymmetric synthesis. Chem Soc Rev, 1996, 25(6): 447–456. DOI: 10.1039/cs9962500447 |
| [5] | Strauss UT, Felfer U, Faber K. Biocatalytic transformation of racemates into chiral building blocks in 100% chemical yield and 100% enantiomeric excess. Tetrahedron Asymmetry, 1999, 10(1): 107–117. DOI: 10.1016/S0957-4166(98)00490-X |
| [6] | Schnell B, Faber K, Kroutil W. Enzymatic racemisation and its application to synthetic biotransformations. Adv Synth Catal, 2003, 345(6/7): 653–666. |
| [7] | Pàmies O, Bäckvall JE. Chemoenzymatic dynamic kinetic resolution. Trends Biotechnol, 2004, 22(3): 130–135. DOI: 10.1016/j.tibtech.2004.01.005 |
| [8] | St.Maurice M, Bearne SL. Hydrophobic nature of the active site of mandelate racemase. Biochemistry, 2004, 43(9): 2524–2532. DOI: 10.1021/bi036207x |
| [9] | Siddiqi F, Bourque JR, Jiang HY, et al. Perturbing the hydrophobic pocket of mandelate racemase to probe phenyl motion during catalysis. Biochemistry, 2005, 44(25): 9013–9021. DOI: 10.1021/bi0473096 |
| [10] | Goriup M, Strauss UT, Felfer U, et al. Substrate spectrum of mandelate racemase:Part 1:variation of the α-hydroxy acid moiety. J Mol Catal B Enzym, 2001, 15(4/6): 207–211. |
| [11] | Narmandakh A, Bearne SL. Purification of recombinant mandelate racemase:improved catalytic activity. Protein Expr Purif, 2010, 69(1): 39–46. DOI: 10.1016/j.pep.2009.06.022 |
| [12] | Resch V, Fabian WMF, Kroutil W. Deracemisation of mandelic acid to optically pure non-natural L-phenylglycine via a redox-neutral biocatalytic cascade. Adv Synth Catal, 2010, 352(6): 993–997. DOI: 10.1002/adsc.200900891 |
| [13] | Fan CW, Xu GC, Ma BD, et al. A novel D-mandelate dehydrogenase used in three-enzyme cascade reaction for highly efficient synthesis of non-natural chiral amino acids. J Biotechnol, 2015, 195: 67–71. DOI: 10.1016/j.jbiotec.2014.10.026 |
| [14] | Nagar M, Bearne SL. An additional role for the br nsted acid-base catalysts of mandelate racemase in transition state stabilization. Biochemistry, 2015, 54(44): 6743–6752. DOI: 10.1021/acs.biochem.5b00982 |
| [15] | Lietzan AD, Nagar M, Pellmann EA, et al. Structure of mandelate racemase with bound intermediate analogues benzohydroxamate and cupferron. Biochemistry, 2012, 51(6): 1160–1170. DOI: 10.1021/bi2018514 |
| [16] | Burley RKM, Bearne SL. Inhibition of mandelate racemase by the substrate-intermediate-product analogue 1, 1-diphenyl-1-hydroxymethylphosphonate. Bioorg Med Chem Lett, 2005, 15(19): 4342–4344. DOI: 10.1016/j.bmcl.2005.06.060 |
| [17] | Bourque JR, Bearne SL. Mutational analysis of the active site flap (20s loop) of mandelate racemase. Biochemistry, 2008, 47(2): 566–578. DOI: 10.1021/bi7015525 |
| [18] | Bearne SL, St.Maurice M, Vaughan MD. An assay for mandelate racemase using high-performance liquid chromatography. Anal Biochem, 1999, 269(2): 332–336. DOI: 10.1006/abio.1999.4018 |
| [19] | Li CX, Jiang XC, Qiu YJ, et al. Identification of a new thermostable and alkali-tolerant α-carbonic anhydrase from Lactobacillus delbrueckii as a biocatalyst for CO2 biomineralization. Bioresour Bioprocess, 2015, 2: 44. DOI: 10.1186/s40643-015-0074-4 |
| [20] | Felfer U, Goriup M, Koegl MF, et al. The substrate spectrum of mandelate racemase:minimum structural requirements for substrates and substrate model. Adv Synth Catal, 2005, 347(7/8): 951–961. |
| [21] | Chen J, Luo XJ, Chen Q, et al. Marked enhancement of Acinetobacter sp.organophosphorus hydrolase activity by a single residue substitution Ile211Ala. Bioresour Bioprocess, 2015, 2: 39. DOI: 10.1186/s40643-015-0067-3 |
| [22] | Yang CC, Ye LD, Gu JL, et al. Directed evolution of mandelate racemase by a novel high-throughput screening method. Appl Microbiol Biotechnol, 2017, 101(3): 1063–1072. DOI: 10.1007/s00253-016-7790-3 |