 2015, Vol. 31
2015, Vol. 31服务
文章信息
- 邓飞龙, 贾先波, 赖松家, 刘益平, 陈仕毅
- Deng Feilong, Jia Xianbo, Lai Songjia, Liu Yiping, Chen Shiyi
- 转录本组装与质量评价
- Transcript assembly and quality assessment
- 生物工程学报, 2015, 31(9): 1271-1278
- Chin J Biotech, 2015, 31(9): 1271-1278
- 10.13345/j.cjb.140508
-
文章历史
- Received: October 24, 2014
- Accepted: January 4, 2015

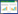




转录组是指某个物种或特定细胞在某一功能状态下的所有RNA总和,包括蛋白质编码RNA和非编码RNA。开展转录组研究能系统分析基因的表达水平及其相互间的调控作用,进一步阐明影响组织发育、性状形成、疾病发生等重要生物学问题的分子机制[1]。转录组研究最初以Sanger测序法和微阵列法为主[2],但随着测序通量和质量的提高,第二代测序技术逐渐成为转录组研究的主流技术[3]。与传统技术相比,第二代测序技术能同时对已知基因和未知基因进行分析,在检测灵敏度上也有显著的提高[1]。
利用第二代测序技术开展转录组研究,主要包括PCR扩增、测序文库构建、序列测定、转录本组装、表达水平定量、基因功能注释等步骤。其中,转录本组装是将在样本处理过程中被随机打断的短片段 (Reads) 拼接还原为完整的转录本,具有较高的技术需求,其组装质量好坏也显著影响着其他分析结果的可靠度,是目前的研究热点和难点[4]。转录本组装方法按照是否依赖于已知的基因组序列,可以分为Genome-guided组装和de novo组装两种方法,它们各具有不同的应用情况与优缺点。因此,系统分析转录本不同组装方法的优缺点以及影响组装质量的关键因素,对我们选择分析软件,提高研究结果的可靠性等方面具有重要的指导意义。
1 转录本组装方法 1.1 Genome-guided 组装Genome-guided组装方法的显著特点是需要提供该物种或近缘物种的基因组序列,首先将测序处理得到的Clean reads比对到该参考基因组上,然后以这些比对上的reads为基础进行转录本的组装 (图1)。该方法的优点在于具有较高的可靠度和灵敏度,但缺点则是必须依赖于已知基因组信息,同时也容易导致假阴性率过高、适用面窄等问题。

|
| 图1 Genome-guided和de novo组装过程 Fig.1 Assembly pipelines of genome-guided and de novo methodologies. |
基于Genome-guided方法的组装软件主要有Cufflinks[5]和Scripture[6],它们在算法与实现上的流程类似,主要可以分为3步:1) 利用短序列比对软件,将测序得到的Clean reads比对到参考基因组上。Cufflinks和Scripture都允许自由选择短序列比对软件,生成SAM或BAM文件格式,并将其作为转录本组装软件的输入。在实际分析流程中,Cufflinks和Scripture都推荐基于bowtie的tophat,或基于bowtie2的tophat2作为该步骤的短序列比对工具[7, 8, 9, 10]。2) 以reads在基因组上的比对信息为依据,确定外显子与外显子间的拼接方式,并以此构建出外显子连接图。当同一条reads的两端序列分别被比对到两个外显子上时,则可以作为将这两个外显子拼接在一起的主要证据。另外,对于两端测序类型 (Paired-end reads) 的配对reads信息也被作为判断两个外显子是否能拼接起来的信号。3) 根据外显子连接图构建转录本。已知所有外显子间的连接信号,即可基于特定的数学模型进行转录本的组装。实际上,Cufflinks与Scripture间最显著的差异也在于使用了不同的数学模型,这也是造成组装结果差异的主要因素。Cufflinks采用二分图模型构建转录本,该方法趋于保守,因而得到的转录本数量会偏少。Scripture采用统计法,即分析连接序列与非连接序列所比对区域的丰度信息,对所有可能的连接路径进行评分,最后依据得分情况选择出可能的转录本。
从Cufflinks和Scripture的算法实现上可以看出,影响转录本组装完整性与可靠性的关键因素是外显子间连接信号的有无以及可靠程度,而它受到测序深度、基因表达丰度、reads比对到基因组上的准确度等因素的影响。因此,当测序得到的reads丰度高、在基因组上的比对位置准确,由此得到的外显子连接信号可保证转录本组装的可靠性。
然而,对低丰度转录本的组装才是难点所在,也最能体现出不同软件间的差异。总体上看,Cufflinks倾向于组装输出更加可靠的转录本,因此在算法上较为保守、在参数控制上更为严格。参数控制上包括过滤相对表达丰度低的转录本 (--min-isoform-fraction),剔除可能是由于前体mRNA不完全剪接留下的内含子序列(--pre-mrna-fraction),丢掉主要由具有多个基因组比对位置的reads所组装出的转录本 (--max-multiread-fraction)。Cufflinks在保证较高准确度的同时,也会导致过高的假阴性率。相比之下,Scripture则倾向于输出更多的转录本,在参数控制中仅能过滤由背景reads构建出来的转录本 (-alpha),因此会带来较高的假阳性率。Cufflinks和Scripture的关键参数与控制命令见表1。
| Assemblers | Parameters | Descriptions | Outputs |
| Cufflinks | --max-multiread-fraction | To set the maximum fraction of reads in one transcript to have multiple locations mapped to genome. (Default: 0.75) | GTF file |
| --min-frags-per-transfrag | To set the minimum number of spliced reads or paired reads to support a transcript. (Default: 10) | ||
| -F | To filter out the minor isoform (s) for a gene with relative abundance (divided by the most abundant isoform) lower than the threshold. (Default: 0.1) | ||
| --trim-3-avgcov-thresh | To trim the 3′ end of transcript when its average coverage depth is lower than the threshold. (Default: 10) | ||
| Scripture | -alpha | To denote the minimum length of transfrag. (Default: 100 bp) | GFF file |
| Velvet-Oases | -cov_cutoff | To remove the contigs with coverage depths lower than the threshold. (Default: 3) | Fasta file |
| -edgeFractionCutoff | At a given node, if edge’s coverage represents less than a given percentage of the sum of coverages of edges coming out of that node, it is removed. (Default: 0.1) | ||
| -min_pair_count | To set the minimum number of spliced reads or paired reads to support the connection between two long contigs. (Default: 4) | ||
| -min_trans_lgth | To set the minimum length of transfrag. (Default: 100 bp) | ||
| Trans-ABySS | --kmer | To set the k-mer size. (Default: 32 bp) | Fasta file |
| Trinity | --min_contig_length | To set the minimum length of assembled transcripts. (Default: 200 bp) | Fasta file |
| --min-kmer-cov | To set the minimum times (frequency) that a k-mer has to be observed. The k-mer with its frequency lower than the threshold will be excluded in the assembly. (Default: 1) | ||
| --trimmomatic | To run the ‘Trimmomatic’ for quality-based trimming of reads. (Default: disabled) | ||
| SOAPdenovo- Trans | -d | To set the minimum times (frequency) that a k-mer has to be observed. Only k-mer with its frequency higher than the threshold will be included in the assembly. (Default: 0) | Fasta file |
| -e | To set the minimum coverage depth for retaining the edges. (Default: 3) | ||
| -k | To set the k-mer size. (Default: min 13 bp, max 31/127 bp) |
De novo组装也称为从头组装,是不依赖于参考基因组的转录本组装方法,即完全利用reads之间的重叠区域信号进行延伸并最终拼接为完整的转录本 (图1)。De novo组装方法的优点是不需要提供参考基因组序列,具有更广的应用,而缺点则在于转录本的完整性差,组装输出转录本的数量会显著上升。
目前基于de novo组装方法的软件主要有Velvet-Oases[11]、Trans-AbySS[12]、Trinity[13]、Rnnotator[14]、SOAPdenovo-Trans[15]等。这些组装软件的实现过程基本一致,包括4大步骤:1) 将测序得到的reads打断为指定长度的k-mer。k-mer的长度参数可以设置为一个固定值,比如Trinity、SOAPdenovo-Trans和Trans-AbySS软件。相反,Velvet-Oases和Rnnotator则采用多个k-mer长度混合组装的方式,即每条reads分多次生成长度不等的k-mer,分别进行转录本的组装,最后对不同结果进行合并。2) 将出现频率最高的k-mer作为种子序列,向两端比对延伸,最终生成不同的Contig 序列。3) 对所有Contig序列寻找重叠区域 (长度为k-1 bp),并构建de Bruijn图。该步骤涉及到较大的计算量,也是影响de novo方法运行速度的关键环节。4) 以de Bruijn图为基础,同时参考reads信息,寻找并构建出所有可能的转录本。
在基于de novo方法的组装软件之间,在对潜在错误序列的处理策略上存在显著差异。SOAPdenovo-Trans是先对低丰度的k-mer进行过滤,然后再构建de Bruijn图。相反,Trans-AbySS和Velvet-Oases是在组装完成后,依据丰度值对转录本进行过滤。而Trinity可以同时在这两个环节进行控制。各软件的关键参数与控制命令见表1。
2 影响转录本组装质量的因素 2.1 PCR 扩增和测序碱基错误第二代测序技术从2005年开始得到快速发展,目前测序准确度已高于99.9%[16]。尽管如此,在单次测序产生数十亿条短序列的规模 下[17],由测序过程本身引起的错误序列也不能被忽略。相比测序错误,PCR扩增过程对序列错误的影响程度会更大。Brodin等[18]表明,在454测序系统中,序列经过60个PCR扩增循环,单个碱基错误率达到0.024%。因此,在转录本组装过程中,由PCR扩增和测序引起的错误碱基是影响组装质量的重要因素。
错误碱基对Genome-guided组装方法的影响主要在于它会降低比对到基因组上的reads的比例,同时也会增加reads被错误比对的概率,而这些错误比对信息会对转录本的组装造成严重的干扰[19, 20]。同样,错误碱基会在de Bruijn图中引入大量的新剪切点,将被用于组装出完全不同的转录本,显著增加错误转录本的数量。相比之下,de novo组装方法对错误碱基会有更高的敏感度[13]。
目前,针对错误碱基的处理,已经开发出了一些特定的算法,包括基于基因组的碱基错误修正算法[21, 22, 23]和基于reads的碱基纠错算 法[19, 24, 25]。虽然依赖这些碱基错误处理算法能在一定程度上减少对转录本组装的影响,但也并不能完全避免该问题的存在。
2.2 测序深度测序深度是指平均每个碱基被测到的频率,主要用于推算所测得的序列在样本中所占的比例。随着测序深度的提高,低丰度表达的基因被测到的概率会增加,从而保证转录本能被完全地组装出来。相反,如果测序深度不足,将导致转录本组装不完整[26]。Genome-guided组装方法一般要求最低测序深度为10×,而de novo组装方法的最低要求为30×[4]。同时,各组装软件在转录本输出时,一般也会依据丰度值进行过滤处理[27]。因此,当测序深度不足时还会导致过多的转录本被错误地过滤丢掉。另外,测序深度过低,能用于支持转录本组装的信息就会较少,导致组装出的转录本的可靠度降低[28]。
需要注意的是,在提高测序深度时,错误碱基的数量也将随之增加,这会加大de novo组装时的干扰信号。因此,在试验设计时,需要考虑将使用的组装方法,在测序深度上取得一个较为合适的平衡点。实践中,解决因单个个体测序深度低而导致转录本组装不完整的问题,可以采用基于多个测序个体混合reads的组装策略[5, 29],但这会显著增加假阳性率。
2.3 重复序列区域以人类基因组 (hg19) 为例,基因组中含有接近50%的重复序列区域[30],而转录组中重复序列区域也高达12.7%[31]。当单一重复区域的长度超过所测得的reads片段长度时,来自该重复区域的reads将可以被比对到多个位置上。因此,Genome-guided组装方法很难对这些在基因组上具有多个比对位置的reads进行区分,无法确定它们的真实来源位置。同样,在de novo组装方法中,这些来自重复区域的reads会导致严重的错误延伸。
对重复区域的组装,de novo组装软件采用倾向组装出尽可能长的contigs的策略,但这只在重复区域长度较短的情况下有效。而在Genome-guided组装方法中,对具有多个比对位置的reads的处理策略可以分为3种:1) 依据短序列比对软件的评分高低,随机选择1个或几个比对位置,用于转录本的组装[6, 32]。2) 考虑满足比对条件的所有位置,均用于转录本的组装[33]。3) 基于相邻的非重复序列区域的丰度值,按照相应的比例将reads分配到不同的比对位置上[5]。相比之下,第3种处理策略具有更好的数学基础,但也没有从根本上解决该问题。
3 转录本组装质量评估从以上的分析可以看出,目前还没有一种算法能够很好地解决转录本组装过程中遇到的众多难题。2009年以来,陆续开发出许多转录本组装软件,它们各有优势,能较好地处理某一或某几方面的问题,但在组装结果的总体质量上均无法得到广泛的认可,这也加大了纯生物学家对软件选择的难度。因此,对转录本组装结果的质量进行评估显得非常重要,但目前还没有完善的比较方法和评价指标体系[4, 11, 34]。
在已有的文献报道中,作者首先根据特定的数学模型去模拟生成已知的reads数据[35, 36, 37],然后再使用相应软件进行组装,将组装得到的结果与模拟数据中的真实值进行比较,从而评价该软件的准确性、完整性和敏感性 (表2)。其中,准确性是指能比对回基因组的转录本的比例。由于样本处理和测序过程的污染,当转录本是由污染序列组装出来的时候,它们则无法被成功比对回基因组上。同时,由于噪音信号的干扰,在组装出的转录本的两端可能存在片段丢失或错误延伸的情况 (完整性)。最后,当片段被正确拼接起来,且两端序列完整并准确时,该转录本即可定义为正确组装的转录本,而该值所占比例即为敏感度。
| Indexes | Formulas | Descriptions |
| Accuracy | ${\rm{ = }}{{{\rm{aNUM}}} \over {{\rm{tSUM}}}}$ | The tSUM is the number of raw transcripts outputted from assemblers. The aNUM, iNUM and sNUM are the number of transcripts mapable to genome, transcripts with observed length equal to expectation and transcripts correctly assembled, respectively. |
| Integrity | ${\rm{ = }}{{{\rm{iNUM}}} \over {{\rm{tSUM}}}}$ | |
| Sensitivity | ${\rm{ = }}{{{\rm{sNUM}}} \over {{\rm{tSUM}}}}$ |
与模拟数据相比,真实转录组数据的复杂程度还远远无法被现有的数学模型所描述。正如前面讨论的一样,也只有当数据结构复杂到一定程度时,软件间的差异才会显现出来。因此,这种简单基于模拟数据比较的评价体系存在明显的缺陷。在分析真实数据时,一般可以采用一些描述性指标,如每个基因含有的平均转录本数目,转录本平均长度、N50长度和N90长度等。然而,这些指标只能用于对组装结果的整体描述,很难进行客观的质量评价。
4 展望转录本的组装质量是决定后续分析结果是否可靠的关键,也是目前在数学模型与算法实现上难度最大的一个环节。基于以上的讨论,我们可以看出Genome-guided和de novo两类组装方法针对不同问题而各有优势,具有一定的互补性。然而,目前还没有能将这两类组装方法进行有效整合的工具或分析流程,尤其是指在算法层面上的整合,这将是一个重要的发展方向。另外,由于转录组数据的高复杂度,采用现有的指标很难对组装质量进行准确可靠的评估。因此,如何开发和建立有效的组装质量评估体系也是该领域面临的一个重要问题。
| [1] | Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet, 2009, 10(1): 57-63. |
| [2] | Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA, 1977, 74(12): 5463-5467. |
| [3] | Metzker Ml. Sequencing technologies—the next generation. Nat Rev Genet, 2009, 11(1): 31-46. |
| [4] | Martin JA, Wang Z. Next-generation transcriptome assembly. Nat Rev Genet, 2011, 12(10): 671-682. |
| [5] | Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc, 2012, 7(3): 562-578. |
| [6] | Guttman M, Garber M, Levin JZ, et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol, 2010, 28(5): 503-510. |
| [7] | Langmead B, Trapnell C, Pop M, et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol, 2009, 10(3): R25. |
| [8] | Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods, 2012, 9(4): 357-359. |
| [9] | Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics, 2009, 25(9): 1105-1111. |
| [10] | Kim D, Pertea G, Trapnell C, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol, 2013, 14(4): R36. |
| [11] | Schulz MH, Zerbino DR, Vingron M, et al. Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics, 2012, 28(8): 1086-1092. |
| [12] | Birol I, Jackman SD, Nielsen CB, et al. De novo transcriptome assembly with ABySS. Bioinformatics, 2009, 25(21): 2872-2877. |
| [13] | Grabherr MG, Haas BJ, Yassour M, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol, 2011, 29(7): 644-652. |
| [14] | Martin J, Bruno VM, Fang Z, et al. Rnnotator: an automated de novo transcriptome assembly pipeline from stranded RNA-Seq reads. BMC Genomics, 2010, 11(1): 663. |
| [15] | Xie Y, Wu G, Tang J, et al. SOAPdenovo-Trans: de novo transcriptome assembly with short RNA-Seq reads. Bioinformatics, 2014, 30: 1660-1666. |
| [16] | Robasky K, Lewis NE, Church GM. The role of replicates for error mitigation in next-generation sequencing. Nat Rev Genet, 2014, 15(1): 56-62. |
| [17] | Ratan A, Miller W, Guillory J, et al. Comparison of sequencing platforms for single nucleotide variant calls in a human sample. PLoS ONE, 2013, 8(2): e55089. |
| [18] | Brodin J, Mild M, Hedskog C, et al. PCR-induced transitions are the major source of error in cleaned ultra-deep pyrosequencing data. PLoS ONE, 2013, 8(7): e70388. |
| [19] | Qu W, Hashimoto SI, Morishita S. Efficient frequency-based de novo short-read clustering for error trimming in next-generation sequencing. Genome Res, 2009, 19(7): 1309-1315. |
| [20] | Dohm JC, Lottaz C, Borodina T, et al. Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Res, 2008, 36(16): e105-e105. |
| [21] | Yang X, Chockalingam SP, Aluru S. A survey of error-correction methods for next-generation sequencing. Brief Bioinform, 2013, 14(1): 56-66. |
| [22] | Yang X, Aluru S, Dorman KS. Repeat-aware modeling and correction of short read errors. BMC Bioinformatics, 2011, 12(Suppl 1): S52. |
| [23] | Wijaya E, Frith MC, Suzuki Y, et al. Recount: expectation maximization based error correction tool for next generation sequencing data. Genome Inform, 2009, 23: 189-201. |
| [24] | Sleep JA, Schreiber AW, Baumann U. Sequencing error correction without a reference genome. BMC Bioinformatics, 2013, 14(1): 367. |
| [25] | Jeck WR, Reinhardt JA, Baltrus DA, et al. Extending assembly of short DNA sequences to handle error. Bioinformatics, 2007, 23(21): 2942-2944. |
| [26] | Wang Y, Ghaffari N, Johnson CD, et al. Evaluation of the coverage and depth of transcriptome by RNA-Seq in chickens. BMC Bioinformatics, 2011, 12(Suppl 10): S5. |
| [27] | Hart T, Komori HK, Lamere S, et al. Finding the active genes in deep RNA-seq gene expression studies. BMC Genomics, 2013, 14(1): 778. |
| [28] | Haas BJ, Chin M, Nusbaum C, et al. How deep is deep enough for RNA-Seq profiling of bacterial transcriptomes. BMC Genomics, 2012, 13(1): 734. |
| [29] | Haas BJ, Papanicolaou A, Yassour M, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc, 2013, 8(8): 1494-1512. |
| [30] | Treangen TJ, Salzberg SL. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet, 2011, 13(1): 36-46. |
| [31] | Kuznetsova IS, Thevasagayam NM, Sridatta Ps, et al. Primary analysis of repeat elements of the Asian seabass (Lates calcarifer) transcriptome and genome. Front Genet, 2014, 5: 223. |
| [32] | Hormozdiari F, Hajirasouliha I, Dao P, et al. Next-generation VariationHunter: combinatorial algorithms for transposon insertion discovery. Bioinformatics, 2010, 26(12): i350-i357. |
| [33] | Mortazavi A, Williams BA, Mccue K, et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods, 2008, 5(7): 621-628. |
| [34] | Steijger T, Abril JF, Engström PG, et al. Assessment of transcript reconstruction methods for RNA-seq. Nat Methods, 2013, 10: 1174-1184. |
| [35] | Hayer K, Pizzaro A, Lahens Nl, et al. Benchmark analysis of algorithms for determining and quantifying full-length mRNA splice forms from RNA-Seq data. BioRxiv, 2014, 007088. |
| [36] | Mcelroy KE, Luciani F, Thomas T. GemSIM: general, error-model based simulator of next-generation sequencing data. BMC Genomics, 2012, 13(1): 74. |
| [37] |
Lu X. Comparison of Transcriptome assembly software for next-generation sequencing technologies[D]. Lanzhou: Lanzhou University, 2013 (in Chinese). 卢戌. 基于第二代测序的转录组组装软件比较研究[D]. 兰州: 兰州大学, 2013. |