 2018, Vol. 58
2018, Vol. 58中国科学院微生物研究所,中国微生物学会,中国菌物学会
文章信息
- 郑智俊, 黄云, 秦楠. 2018
- Zhijun Zheng, Yun Huang, Nan Qin. 2018
- 人体肠道宏基因组生物信息分析方法
- A review of bioinformatic methodologies in metagenomics
- 微生物学报, 58(11): 2020-2032
- Acta Microbiologica Sinica, 58(11): 2020-2032
-
文章历史
- 收稿日期:2018-03-15
- 修回日期:2018-07-24
- 网络出版日期:2018-07-30

引用本文 |



2. 同济大学附属第十人民医院, 上海 200072
2. Shanghai Tenth People's Hospital Affiliated to Tongji University, Shanghai 200072, China
肠道是含有微生物物种数量最多、功能最全面的微生态群落器官,据报道,人类肠道中有多达39万亿个细菌细胞,是人类细胞数量的1.3倍,而且这些细菌编码的基因数量是人类基因的100多倍[1]。人体肠道微生物的定殖显著影响人类的生理和营养,对人们生活至关重要。先前研究已经表明肠道微生物有助于从食物中获取能量,调节免疫力、体内激素平衡、中枢神经系统成熟以及行为特征。此外,肠道微生物组的变化也与人类多种疾病相关,如肥胖[2-5]、代谢综合征[6]、类风湿性关节炎[7]、炎症性肠病[8-9]、阿尔茨海默病[10]、糖尿病[11]、流感病毒感染[12]、症状性动脉粥样硬化[13]和肝脏疾病[14-17]。
要了解肠道微生物对人类健康的影响,需阐明肠道微环境的组成、多样性和功能[1]。基于DNA的测序分析克服了传统微生物培养技术的局限性,并取得了新的进展,极大地扩大了目前对肠道微生物多样性的认识[18]。由美国国立卫生研究院启动的人类微生物组计划从300个美国人的不同人体部位产生了大量16S rRNA基因数据集,其中显示人类肠道微生物群主要由2个门主导,拟杆菌门和厚壁菌门。然而,16S rRNA基因基于扩增片段的分析只能检测到属和以上级别的微生物,这意味着分类学多样性与功能将在种及其以下水平(即亚种和株)被忽略。宏基因组测序的目标是整个基因组DNA序列,体现了替代16S rRNA基因测序的强大策略,用于分析复杂的微生物群落。宏基因组学研究已应用于人类肠道微生物群,以产生大量可用于表征微生物群落的组成和功能的数据[19]。更重要的是,基于全基因组DNA的测序可以检测物种水平的微生物,并且比基于16S rRNA基因的方法更适合于肠道微生物组和人类疾病之间的关联研究。
1 宏基因组生物信息分析方法 1.1 物种注释宏基因组物种注释也叫宏基因组物种比对,是将经过拆分、质控、去宿主后的高质量clean reads,与参考基因组或marker基因做比对。宏基因组物种注释的参考微生物包括细菌、古细菌、病毒和真菌四大类,它们都来源于当前流行的各大数据库,包括NCBI数据库(http://www.ncbi.nlm.nih.gov/genome)、人体微生物计划(http://www.hmpdacc.org)、GOLD数据库(https://gold.jgi.doe.gov)、酵母基因组数据库(http://www.yeastgenome.org)、真菌基因组行动(http://www.broadinstitute.org)、人类口腔微生物数据库(http://www.homd.org/)、真菌数据库(http://fungidb.org)。随着生物信息分析行业的发展,越来越多好的软件被开发出来,比对软件只是其中一小类,常用短序列比对软件包括:SOAP[20]、BWA[21]、Bowtie[22]、Martin[23]等。有人使用了RefCov (http://gmt.genome.wustl.edu/gmt-refcov)软件,对6种常见短序列比对软件做了评估,主要通过统计它们的覆盖广度和覆盖深度。这项研究表明,这些比对软件都可以在肠道宏基因组数据中发挥各自的价值,其中CLC aligner软件的计算时间最短,使用的内存最少。宏基因组物种注释还需要基于比对结果,获得相应的物种分类信息。虽然现在可用的数据库很多,但是环境中还有许多微生物分类信息没有收录在参考数据库中。目前有少量软件既可以比对微生物基因组,又可以对微生物分类,还可以统计微生物丰度,例如MetaPhlAn2[24]软件、Kraken[10]软件、Genometa[25]软件。其中Kraken软件是基于Kmer的策略,能非常快速、准确找出复杂环境中的微生物基因组,并且物种丰度计算的速度比Metaphlan2软件快11倍多,其核心是Kraken有一种特殊数据库,用以预先计算基因组中包含的特殊Kmer序列,而Genometa有很好的交互性。
1.2 肠型2011年,肠型(Enterotypes)的概念首次在《自然》杂志上由Arumugam等[26]提出,该研究发现可以将人类肠道微生物组分成稳定的3种类型,因为这3种类型不受年龄、性别、体重以及地域限制,表现出较高的稳定性,与血型具有很高的相似性,所以将其定义为肠型。2012年,在其他灵长类动物中也发现了肠型的存在[27]。但是由于肠型并不是完全离散的分布,有研究发现在人肠道微生物组中存在4种可互相转换的肠型[28],另一项针对美国人群的研究则报道称发现了2种肠型,这些结果也引起了国际上对于肠型概念的广泛讨论与争议。
肠型是肠道微生物组在高维数据空间中呈现出的客观存在的聚集效应。目前流行的肠型计算方法有2种,一种是基于样品间的Jensen-Shannon距离,利用围绕中心点划分算法(PAM)进行聚类,最佳分类数目通过Calinski-Harabasz (CH)指数确定[29];另一种则是直接基于物种丰度,利用狄利克雷多项混合模型(DMM)进行肠型分类[30]。之后,肠型的可视化通过使用R软件的‘ade4’包的类间分析、主成分分析或主坐标分析构建图形来实现[31]。由于在样品中某些属或种的相对丰度很低,所以在分析之前,可以用适当的阈值来过滤相对丰度较低的物种以降低噪声。
根据物种平均相对丰度大于0.1%过滤后的肠型分类可视化,如图 1所示。

|
| 图 1 肠型示意图 Figure 1 An example of Enterotypes in the human gut microbiome. This figure was plotted using our own data in Principal Coordinates Analysis. |
生物信息学工具已被广泛用于肠型的研究,相比与血型一样的离散类型,对肠型分布更好的理解应该是在物种特征的高维空间中的密集分布(就如http://enterotype.embl.de/所描述的:Enterotypes are not discrete types,like blood types,but rather densely populated regions in a higher dimensional space of microbiome features),因此将肠型作为根据肠道微生物组对个体进行分层的标准或许将更为合适。
最新的《自然∙微生物学》杂志报道了学界对于肠型的最新认识,调解了之前关于肠型的一些争论[32]。首先,肯定了肠道菌群大数据在人群中成层现象(population stratification)的客观存在性[32];其次,对于之前出现或2种肠型、或3种肠型、或4种肠型的争论进行了系统分析,发现随着大数据的积累,肠型必然是连续的而非离散的,并且研究了各种不同方法对结果的影响,发现造成差异的原因是由于不同方法抽取不同维度进行观测,观测的其实都是复杂肠道菌群大数据整体的某个子集的不同切面[32]。然而不论如何观测,以后更多数据积累发现新类型,都无法否认目前人群中常见的几种肠型,如Bacteroides型、Prevotella型、Firmicutes型等的客观存在性。
1.3 分类多样性的描述在宏基因组学中,分类多样性通常表现为阿尔法(α)多样性和贝塔(β)多样性,这是生态学研究中常用的术语。
阿尔法多样性:单个样品中的分类群数量和这些群体的平均丰度称为阿尔法多样性,可以使用统计学方法确定,如Shannon-Wiener指数[33]、系统发育多样性全树分析[34]和Simpson指数[35]。通常,这些指数是逐步计算和递归计算的,以确定采样数据量是否具有足够的代表性以形成稀疏曲线。
贝塔多样性:阿尔法多样性描述了单个样本的特性,而贝塔多样性描述了样本之间的相似性。对于贝塔多样性分析,样品之间的距离/相似性,往往使用例如Bray-Curtis测量[36-37]方法和加权/未加权的UniFrac[1, 24, 38-40]方法。样本之间的相似性可以使用精准算法在二维图中可视化[41]。样本组成可以看作是一个向量,因此可以看作是存在于多维坐标系中的一个点。协调系统目录的简单和转换表明样本之间的距离,这是主成分分析的精确性质。
1.4 组装和基因集构建因为二代测序技术得到的reads仍旧较短,且其中的微生物组装复杂,因而基于组装得到相对更长的contigs进行的下游分析将更有效。由于在宏基因组中超过90%的微生物是未知的[42],不依赖于参考序列的从头组装就显得很有必要了。现有的宏基因组组装算法有很多,绝大部分都是基于de Brujin图构建的方法,例如,MetaVelvet[43]和Meta-IBDA[44]通过reads构建de Brujin图,并根据de Brujin图的特性识别代表基因组特异性的子图(sub-graphs);Genovo[45]根据组装概率模型识别最大可能的序列重构,从而得到具有最大似然值的contigs集合。Vollmers[46]对这些组装软件进行了评测,并提出了最优组装软件的选择依赖于样品的类型、可获得的计算资源,以及更重要的是研究目的。
通过基因预测可以识别宏基因组中的基因编码序列,从而了解宏基因组样品中微生物的基因功能。常用的基因预测软件有MetaGeneMark[47]、Glimmer-MG[48]、MetaGene[49]、Orphelia[50]、FragGeneScan[51]和MetaGun[52]。Trimble等[53]通过统计模拟数据对这些预测方法进行了比较,发现这些方法的性能会根据reads的性质(例如测序错误率和reads长度等)而有所变化,并且不同的方法将根据不同性质的阈值产生最优准确性,因此建议研究者需要基于自身研究数据的特性来选择合适的方法以及对应的参数。
当对多个宏基因组样品进行比较分析的时候,我们需要先根据每个样品预测得到的基因构建一个去冗余的基因集。我们通常使用cd-hit[54]进行去冗余基因组的构建,首先,将预测的基因序列进行两两比对,然后根据比对结果将能比对到另外一条较长序列(相似度大于90%且覆盖度大于95%)的基因序列列为冗余序列去除,最终得到一个去冗余的基因集。考虑到由于测序数据有限会导致一些低丰度微生物不能被检测到,所以通过整合已经公布的基因集,例如MetaHIT基因集[55]、HMP基因集合[56]和LC基因集[14]等,将有利于优化自身数据分析得到的基因集合。
1.5 差异分析差异基因分析(或差异物种分析)就是在基因丰度表(或物种丰度表)中寻找针对不同组之间有显著性差异的一种分析,一般使用针对非正态分布的非参检验方法,具体而言,有Wilcoxon秩和检验、Mann-Whitney U检验和Wilcoxon signed-rank检验等。其中,对于2个独立样本(两组来源于不同总体)的差异分析方法可以使用Wilcoxon秩和检验与Mann-Whitney U检验,Wilcoxon秩和检验与Mann-Whitney U检验并没有实质性的差别。对于2个配对样本(两组来源于同一个总体),应该使用Wilcoxon signed-rank检验来代替Mann-Whitney U检验(或Wilcoxon秩和检验)。当样本量很小时,我们采用Wilcoxon rank-sum permutation检验,在这种情况下,P值是基于置换(排列)的估计分布来代替一个固定的正态分布来计算的。对于3组或以上的差异基因检验方法有Kruskal-Wallis检验、Friendman检验等。
通常的方法是,在每组中过滤掉平均丰度低于10-8的基因,然后用差异检验方法来鉴定与实验变量相关的基因。因为做差异基因的筛选,有多个基因要筛选,需要做多重假设检验,通过检验所获得的P值直接与设定的显著性水平进行比较会有很大的误差,所以我们通过多重假设检验,将所获得的P值通过FDR (false discovery rate)来进行校正,并通常设置校正后的阈值(Q值)为0.05。
1.6 丰度表的计算物种丰度是指物种数量的多少。肠道的物种丰度表为肠型分析、肠道多样性计算以及寻找疾病的微生物标记物奠定了基础。
目前,有两种较为常用的计算物种多样性的方法。一种是将测序所得到的宏基因组序列与全基因组序列进行比对,经计算后得到物种丰度表。该计算方法将唯一比对到的序列丰度与多重比对到的序列丰度求和得到物种丰度[14],按照公式(1-4)计算。

|
公式(1) |
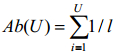
|
公式(2) |
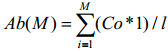
|
公式(3) |

|
公式(4) |
式中,Ab(S)表示S物种的物种丰度,U表示唯一比对到S物种上的序列数量,M表示比对到多个物种的序列数量,l表示参考基因组的长度,N表示物种的数量,若物种S的Ab(U)为0,那么,Co也为0。在计算完成所有物种丰度之后,将物种S的物种丰度除以所有物种丰度之和,就可以得到某样本的物种丰度表。
另一种方法是用MetaPhlAn2[24]或mOTU[39]软件将测序得到的宏基因组序列比对到标记序列数据库上,计算后就可以得到物种丰度表。mOTU和MetaPhlAn2比对的数据库有所不同,MetaPhlAn2是将序列比对到一个已构建好的肠道数据库上,该数据库包含了13500多个细菌古菌,3500个病毒和110个真菌的序列标记物,将比对到物种S的标记序列上的序列数除以比对到标记序列上的序列总数,就得到了物种S的物种丰度,计算所有的物种丰度就构成了物种丰度表,mOTU计算物种丰度需要依赖于40个通用单拷贝系统发育标记基因序列,这些基因在绝大多数已知生物的基因组中以单拷贝形式出现,将测序序列比对到这些标记基因可以生成物种丰度表,这40个标记基因都是从3445个完整的细菌基因组和来自263个公开的宏基因组的未知物种中提取得到的。
基因丰度表是寻找差异基因、研究疾病与健康差异、挖掘深层生物机理的基础。在计算基因丰度之前,需要先构建非冗余基因集,构建好基因集之后就可以将原始序列用SOAPaligner[20]、BLAST[57]、BLAT[58]、BWA[21]或Bowtie2[32]等比对软件比对到基因集上,通过计算可以得到基因丰度。计算方法与计算物种丰度类似,假设现有基因G,基因G的丰度就等于唯一比对到的序列丰度与多重比对到的序列丰度之和。按照公式(5-8)计算。
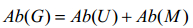
|
公式(5) |

|
公式(6) |
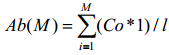
|
公式(7) |
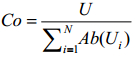
|
公式(8) |
式中,Ab(G)表示基因G的基因丰度,U表示唯一比对到某基因上的序列数量,M表示比对到多个基因的序列数量,l表示基因序列的长度,N表示基因集中的数量,若基因G的Ab(U)为0,那么,Co也为0。在计算完成所有基因丰度之后,将基因G的基因丰度除以所有的基因丰度之和,就得到某样本的基因丰度表。
1.7 功能分析目前,功能分析的应用非常普遍,以代谢信号通路为例,与其相关的文献逐年增加[59-61],所以宏基因组功能分析将会是一个非常普遍而且重要的分析内容。宏基因组功能分析可以获得基因的注释信息,了解相关的代谢信号通路,有助于研究者从系统水平去了解基因的生物学功能,进一步揭示宿主与菌群之间的关系[60]。
宏基因组功能分析的主要参考数据库包括京都基因与基因组百科全书(KEGG)数据库[62]、EggNOG数据库[63]、GO数据库[64]、耐药基因(ARDB)数据库[65]、碳水化合物酶(CAZY)数据库[66]、群体感应(QSDB)数据库、病原与宿主互作(PHI)数据库[67]、病原菌毒力因子(VFDB)数据库[68]、转运蛋白分类(TCDB)数据库[69]等。
代谢信号通路(pathway)的主要参考数据库有KEGG和EggNOG。KEGG是基因组破译方面的数据库,是一个整合了基因组、化学和系统功能信息的数据库[62],包括完整和部分测序的基因组序列,更高级的功能信息存储在PATHWAY数据库里,包括图解的细胞生化过程如代谢、膜转运、信号传递、细胞周期,还包括同系保守的子通路等信息[70]。KEGG通过图形方式呈现众多的代谢途径以及各途径之间的关系,这样可以使研究者能够对其所要研究的代谢途径有一个直观全面的了解。
EggNOG数据库是利用Smith-Waterman比对算法对构建的基因直系同源类群进行了功能描述和功能分类的注释,从另一个角度提供基因功能分析。
与KEGG和EggNOG存在一定差异的是,GO数据库主要用于研究基因功能,而非研究基因和蛋白功能。
其他数据库更多专注于具有特殊功能的基因,如CAZy数据库包含了碳水化合物酶类的物种来源、酶功能EC分类、基因序列、蛋白质序列及其结构等信息。
ARDB数据库包含细菌病原菌的多种抗性基因数据,通过该数据库的注释可以找到耐药性相关的基因名称以及所耐受的抗生素的种类。
PHI数据库包含了被实验证实对病原体与宿主相互作用有影响的基因,主要来源于真菌、卵菌和细菌病原体,宿主包括动物、植物和真菌。
TCDB数据库是一个用于运输蛋白质研究的参考数据,包含了各种生物体运输系统有关蛋白的序列、结构、功能以及进化信息。
VFDB数据库包含细菌病原体的毒力基因序列信息,被广泛应用于毒力因子基因鉴定。
通过序列比对,将基因集比对到相应的参考数据库,获得基因的注释信息,对注释信息分析,了解基因参与的生物学功能。通常使用BLAST[57]软件进行基因集注释的序列比对。
1.8 基因之间的关联分析和MGS (MetaGenomic Species)基因之间的关联分析与β多样性分析相似,通常采用斯皮尔曼等级相关系数和皮尔逊相关系数计算相关性。当2个物种之间的相关系数高时,它可能表明这些物种是相关的。同样地,当宏基因组分析中2个基因之间的相关系数高时,可以表明这些基因来自相同的物种。
在宏基因组分析中,可以根据它们的相关系数将基因分组在一起。当选择合适的分组标准时,来自相同物种的基因很可能被聚集在一起,这样的基因群被称为MGS,并且这些基因群中的基因可以与目前可用的参考基因组比对以验证“物种”检测的分组方法,一些未比对上的基因群也许来源于未知物种。从MGS数据中可以鉴定和提取属于特定物种的基因。因此,微生物基因组可以使用宏基因组数据进行组装,MGS可以提供可用于计算分类组成的潜在参考基因组。
1.9 CAG (co-abundance gene group)方法可用于未知物种鉴定CAG方法是2014年在《自然∙生物技术》杂志上报道出来的人体肠道宏基因组数据分析方法[13]。这种方法的思想和MGS的思想有一定相同之处,都利用了这样一种思想或理论:在同一个株或种中的基因,这些基因的丰度在群体中具有丰度一致性。为了解决使用MGS方法时仅关注基因标记的局限性,canopy算法被用来鉴定共丰富基因簇(CAG),CAG分析基于基因丰度鉴定物种,并且每个CAG可以被看作是部分微生物或完整微生物。
当基因丰度表包含足够数量的样本时,可以应用canopy算法来识别CAG。canopy算法使用Pearson相关系数和Spearman相关系数作为阈值,在对只有1个基因的canopies进行第一轮聚类和过滤之后,根据簇的平均丰度,使用Pearson相关系数作为阈值再次被应用到canopy算法中,第二轮簇可以包含重叠基因。因此,对于出现在多于1个簇中的基因,基因及其相关簇之间的距离能够被确定,对于每个重叠基因,选择最近的簇。最后,选择含有超过700个基因的簇最可能是含有细菌基因组部分的CAG。
基于CAG分析的结果,可以在人肠道微生物群中鉴定出可能显著影响宿主的未分类或难以理解的微生物(也称为生物暗物质)。在过去的研究中,生物暗物质一般被忽略,因此,许多疾病相关的肠道微生物可能未被检测到。CAG中的重叠群和基因可以产生无法从当前的细菌基因组数据库获得的大量新信息。
1.10 医学分类器医学分类器是通过特征工程中的特征选择的方法挑选出若干不同级别的bio-markers,并根据bio-markers来构建的一种分类模型,它采用分类评价指标来评估模型效果的好坏,目的是用于识别疾病与健康人群。
医学分类器是来自机器学习中的分类算法,包括Logistictic回归、Support Vector Machine (SVM)、Random Forest、人工神经网络、朴素贝叶斯等。除了分类算法,特征选择对分类性能也会有直接的影响,特征选择就是在我们获得的给定特征集中选择出与分类相关的特征子集的过程。mRMR (maximal relevance and minimal redundancy)是一种常用的特征选择的评估准则。评估方法一般包括Filter、Wrapper、Hybrid、Embeded,其中Wrapper是较为传统和常用的方法,它是直接把最终将要使用的学习器的性能作为特征子集的评价准则。通过分类算法得到的分类器的性能最终是由分类评价指标来确定,分类评价指标是用来评价分类器分类性能好坏的一种指标,常见的评价指标有正确率、错误率、灵敏度、特异度、精度、Matthews correlation coefficient (MCC)、Receiver operating characteristic (ROC)。其中MCC和ROC不受分组样本数量影响;ROC是根据一系列不同的二分类方式,以真阳性率(灵敏度)为纵坐标,假阳性率(1-特异度)为横坐标绘制的曲线,直观描绘了分类器在TP和FP间的trade-off,AUC (area under curve)是ROC曲线下方面积之和,它以数值的形式来评估分类器的好坏,AUC的取值范围在(0.5, 1.0)范围内,AUC的值越大,分类的性能就越好。若分类评价指标最终显示分类器性能不佳,则需重新构建分类器直至分类性能达到稳定为止。
医学分类器的引入对于构建用于临床诊断的基于肠道微生物的非接触检测方法具有重要意义。
2 展望随着生物信息行业的迅猛发展,越来越多的组装软件被开发出来,然而还有许多问题有待解决。从人体肠道宏基因组测序数据进行全基因组组装,为下一步基因预测、基因集构建、基因丰度统计提供了重要的基础数据。肠道菌群非常复杂,许多不同菌株来自相同物种,以及许多不同物种来自相同属,这些高度相似DNA序列会混淆组装结果,导致不正确的基因组序列出现,因此组装算法还有很大的提升空间。二代测序的短读长限制了contig的长度,随着三代测序技术成熟,基于三代PacBio的SMRT单分子测序在宏基因组方面也已开展了一些应用,PacBio产生的序列平均长度在5-50 kb,其中50%左右的序列长度大于14 kb,宏基因组拼接组装效果有了很大程度的提高,能组装出更长的contig,提高了短reads的使用率,增加了完整基因的数量,这表明宏基因组的组装又上升了一个台阶。随着测序读长不断增加,测序质量的不断提高,将三代测序数据应用到宏基因组中将是未来研究的一大方向。
随着技术的发展,亚种或株水平的研究必然对人体肠道宏基因组领域产生深刻影响。大量(60%-70%)的肠道微生物目前还无法从现有国际数据库中找到参考基因组,虽然人体肠道宏基因组领域有CAG等方法对亚种或株通过生物信息学方法进行分析,但对这个方向的根本性突破还是需要准确度更高、读长更长的测序技术如三代测序技术去推动。当测序准确度提高、测序读长接近微生物自身基因组长度,大量未知的肠道微生物基因组将通过最简单、直接的方式整体性地被呈现出来,所谓的生物暗物质将向我们揭开神秘面纱。
2017年,美国克利夫兰医学中心将“利用微生物组预防、诊断和治疗疾病”列为2017年十大医疗科技创新之首。微生物组技术本身就是医疗前沿创新技术,随着近年来各项新技术的发展,微生物组与多个热门领域新技术相结合,更体现出了微生物组技术的活力和未来的广阔应用价值。从2017年到2018年,在肿瘤免疫治疗领域,高水平学术期刊《科学》连续报道了肠道菌群显著影响PD-1 (程序性死亡受体1)药物治疗的效果[71-72],并且文章通过宏基因组技术找到与PD-1免疫疗法相关的10种微生物。在未来肿瘤免疫治疗用药方案选择前,通过肠道菌群检测及宏基因组分析或是一种常规的手段。而另一个炙手可热的领域CRISPR/Cas9基因编辑技术,在未来改造菌群对抗日益严峻的抗生素滥用、细菌耐药性乃至通过菌群改造治疗疾病,也有与宏基因组技术相结合应用的广阔空间。
致谢:感谢吴春燕、范芳芳、宋文峰、李心如、马圣、熊潇、马玮在资料收集和文章撰写中提供了帮助。
| [1] | Sender R, Fuchs S, Milo R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biology, 2016, 14(8): e1002533. DOI:10.1371/journal.pbio.1002533 |
| [2] | Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: Human gut microbes associated with obesity. Nature, 2006, 444(7122): 1022-1023. DOI:10.1038/4441022a |
| [3] | Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, Egholm M, Henrissat B, Heath AC, Knight R, Gordon JI. A core gut microbiome in obese and lean twins. Nature, 2009, 457(7228): 480-484. DOI:10.1038/nature07540 |
| [4] | Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature, 2006, 444(7122): 1027-1031. DOI:10.1038/nature05414 |
| [5] | Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proceedings of the National Academy of Sciences of the United States of America, 2005, 102(31): 11070-11075. DOI:10.1073/pnas.0504978102 |
| [6] | Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman SV, Knight R, Ley RE, Gewirtz AT. Metabolic syndrome and altered gut microbiota in mice lacking toll-like receptor 5. Science, 2010, 328(5975): 228-231. DOI:10.1126/science.1179721 |
| [7] | Zhang X, Zhang DY, Jia HJ, Feng Q, Wang DH, Liang D, Wu XN, Li JH, Tang LQ, Li Y, Lan Z, Chen B, Li YL, Zhong HZ, Xie HL, Jie ZY, Chen WN, Tang SM, Xu XQ, Wang XK, Cai XH, Liu S, Xia Y, Li JY, Qiao XY, Al-Aama JY, Chen H, Wang L, Wu QJ, Zhang FC, Zheng WJ, Li YZ, Zhang MR, Luo GW, Xue WB, Xiao L, Li J, Chen WT, Xu X, Yin Y, Yang HM, Wang J, Kristiansen K, Liu L, Li T, Huang QC, Li YR, Wang J. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nature Medicine, 2015, 21(8): 895-905. DOI:10.1038/nm.3914 |
| [8] | Lepage P, Häsler R, Spehlmann ME, Rehman A, Zvirbliene A, Begun A, Ott S, Kupcinskas L, Doré J, Raedler A, Schreiber S. Twin study indicates loss of interaction between microbiota and mucosa of patients with ulcerative colitis. Gastroenterology, 2011, 141(1): 227-236. DOI:10.1053/j.gastro.2011.04.011 |
| [9] | Garrett WS, Gallini CA, Yatsunenko T, Michaud M, Dubois A, Delaney ML, Punit S, Karlsson M, Bry L, Glickman JN, Gordon JI, Onderdonk AB, Glimcher LH. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host & Microbe, 2010, 8(3): 292-300. |
| [10] | Hill JM, Bhattacharjee S, Pogue AI, Lukiw WJ. The gastrointestinal tract microbiome and potential link to Alzheimer's disease. Frontiers in Neurology, 2014, 5: 43. |
| [11] | Qin JJ, Li YR, Cai ZM, Li SH, Zhu JF, Zhang F, Liang SS, Zhang WW, Guan YL, Shen DQ, Peng YQ, Zhang DY, Jie ZY, Wu WX, Qin YW, Xue WB, Li JH, Han LC, Lu DH, Wu PX, Dai YL, Sun XJ, Li ZS, Tang AF, Zhong SL, Li XP, Chen WN, Xu R, Wang MB, Feng Q, Gong MH, Yu J, Zhang YY, Zhang M, Hansen T, Sanchez G, Raes J, Falony G, Okuda S, Almeida M, LeChatelier E, Renault P, Pons N, Batto JM, Zhang ZX, Chen H, Yang RF, Zheng WM, Li SG, Yang HM, Wang J, Ehrlich SD, Nielsen R, Pedersen O, Kristiansen K, Wang J. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature, 2012, 490(7418): 55-60. DOI:10.1038/nature11450 |
| [12] | Qin N, Zheng BW, Yao J, Guo LH, Zuo J, Wu LJ, Zhou JW, Liu L, Guo J, Ni SJ, Li A, Zhu YX, Liang WF, Xiao YH, Ehrlich SD, Li LJ. Influence of H7N9 virus infection and associated treatment on human gut microbiota. Scientific Reports, 2015, 5: 14771. DOI:10.1038/srep14771 |
| [13] | Karlsson FH, F k F, Nookaew I, Tremaroli V, Fagerberg B, Petranovic D, Bäckhed F, Nielsen J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nature Communications, 2012, 3: 1245. DOI:10.1038/ncomms2266 |
| [14] | Qin N, Yang FL, Li A, Prifti E, Chen YF, Shao L, Guo J, Le Chatelier E, Yao J, Wu LJ, Zhou JW, Ni SJ, Liu L, Pons N, Batto JM, Kennedy SP, Leonard P, Yuan CH, Ding WC, Chen YT, Hu XJ, Zheng BW, Qian GR, Xu W, Ehrlich SD, Zheng SS, Li LJ. Alterations of the human gut microbiome in liver cirrhosis. Nature, 2014, 513(7516): 59-64. DOI:10.1038/nature13568 |
| [15] | Benten D, Wiest R. Gut microbiome and intestinal barrier failure-The "Achilles heel" in hepatology?. Journal of Hepatology, 2012, 56(6): 1221-1223. DOI:10.1016/j.jhep.2012.03.003 |
| [16] | Yan AW, Fouts DE, Brandl J, Stärkel P, Torralba M, Schott E, Tsukamoto H, Nelson KE, Brenner DA, Schnabl B. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology, 2011, 53(1): 96-105. DOI:10.1002/hep.24018 |
| [17] | De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, Collini S, Pieraccini G, Lionetti P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proceedings of the National Academy of Sciences of the United States of America, 2010, 107(33): 14691-14696. DOI:10.1073/pnas.1005963107 |
| [18] | Cho I, Blaser MJ. The human microbiome: at the interface of health and disease. Nature Reviews Genetics, 2012, 13(4): 260-270. DOI:10.1038/nrg3182 |
| [19] | Sender R, Fuchs S, Milo R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biology, 2016, 14(8): e1002533. DOI:10.1371/journal.pbio.1002533 |
| [20] | Li R, Li Y, Kristiansen K, Wang J. SOAP: short oligonucleotide alignment program. Bioinformatics, 2008, 24(5): 713-714. DOI:10.1093/bioinformatics/btn025 |
| [21] | Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 2009, 25(14): 1754-1760. DOI:10.1093/bioinformatics/btp324 |
| [22] | Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology, 2009, 10(3): R25. DOI:10.1186/gb-2009-10-3-r25 |
| [23] | Martin J, Sykes S, Young S, Kota K, Sanka R, Sheth N, Orvis J, Sodergren E, Wang ZY, Weinstock GM, Mitreva M. Optimizing read mapping to reference genomes to determine composition and species prevalence in microbial communities. PLoS One, 2012, 7(6): e3642. |
| [24] | Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, Tett A, Huttenhower C, Segata N. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nature Methods, 2015, 12(10): 902-903. DOI:10.1038/nmeth.3589 |
| [25] | Davenport CF, Neugebauer J, Beckmann N, Friedrich B, Kameri B, Kokott S, Paetow M, Siekmann B, Wieding-Drewes M, Wienhöfer M, Wolf S, Tümmler B, Ahlers V, Sprengel F. Genometa--a fast and accurate classifier for short metagenomic shotgun reads. PLoS One, 2012, 7(8): e41224. DOI:10.1371/journal.pone.0041224 |
| [26] | Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto JM, Bertalan M, Borruel N, Casellas F, Fernandez L, Gautier L, Hansen T, Hattori M, Hayashi T, Kleerebezem M, Kurokawa K, Leclerc M, Levenez F, Manichanh C, Nielsen HB, Nielsen T, Pons N, Poulain J, Qin JJ, Sicheritz-Ponten T, Tims S, Torrents D, Ugarte E, Zoetendal EG, Wang J, Guarner F, Pedersen O, de Vos WM, Brunak S, Doré J, MetaHIT Consortium (additional members), Weissenbach J, Ehrlich SD, Bork P. Enterotypes of the human gut microbiome. Nature, 2011, 473(7346): 174-180. DOI:10.1038/nature09944 |
| [27] | Moeller AH, Degnan PH, Pusey AE, Wilson ML, Hahn BH, Ochman H. Chimpanzees and humans harbour compositionally similar gut enterotypes. Nature Communications, 2012, 3: 1179. DOI:10.1038/ncomms2159 |
| [28] | Ding T, Schloss PD. Dynamics and associations of microbial community types across the human body. Nature, 2014, 509(7500): 357-360. DOI:10.1038/nature13178 |
| [29] | Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, Bewtra M, Knights D, Walters WA, Knight R, Sinha R, Gilroy E, Gupta K, Baldassano R, Nessel L, Li H, Bushman FD, Lewis JD. Linking long-term dietary patterns with gut microbial enterotypes. Science, 2011, 334(6052): 105-108. DOI:10.1126/science.1208344 |
| [30] | Holmes I, Harris K, Quince C. Dirichlet multinomial mixtures: generative models for microbial metagenomics. PLoS One, 2012, 7(2): e30126. DOI:10.1371/journal.pone.0030126 |
| [31] | Dray S, Dufour AB. The ade4 package: Implementing the duality diagram for ecologists. Journal of Statistical Software, 2007, 22(4): 1-20. |
| [32] | Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature Methods, 2012, 9(4): 357-359. DOI:10.1038/nmeth.1923 |
| [33] | Spellerberg IF, Fedor PJ. A tribute to Claude Shannon (1916-2001) and a plea for more rigorous use of species richness, species diversity and the 'Shannon-Wiener' index. Global Ecology and Biogeography, 2003, 12(3): 177-179. DOI:10.1046/j.1466-822X.2003.00015.x |
| [34] | Schimleck LR, Kube PD, Raymond CA, Michell AJ, French J. Estimation of whole-tree kraft pulp yield of Eucalyptus nitens using near-infrared spectra collected from increment cores. Canadian Journal of Forest Research, 2005, 35(12): 2797-2805. DOI:10.1139/x05-193 |
| [35] | Hunter PR, Gaston MA. Numerical index of the discriminatory ability of typing systems: an application of Simpson's index of diversity. Journal of Clinical Microbiology, 1988, 26(11): 2465-2466. |
| [36] | Clarke KR, Somerfield PJ, Chapman MG. On resemblance measures for ecological studies, including taxonomic dissimilarities and a zero-adjusted Bray-Curtis coefficient for denuded assemblages. Journal of Experimental Marine Biology and Ecology, 2006, 330(1): 55-80. DOI:10.1016/j.jembe.2005.12.017 |
| [37] | Beals EW. Bray-Curtis ordination: an effective strategy for analysis of multivariate ecological data. Advances in Ecological Research, 1984, 14: 1-55. DOI:10.1016/S0065-2504(08)60168-3 |
| [38] | Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, Heath AC, Warner B, Reeder J, Kuczynski J, Gregory GJ, Lozupone CA, Lauber C, Clemente JC, Knights D, Knight R, Gordon JI. Human gut microbiome viewed across age and geography. Nature, 2012, 486(7402): 222-227. DOI:10.1038/nature11053 |
| [39] | Kultima JR, Sunagawa S, Li JH, Chen WN, Chen H, Mende DR, Arumugam M, Pan Q, Liu BH, Qin JJ, Wang J, Bork P. MOCAT: a metagenomics assembly and gene prediction toolkit. PLoS One, 2012, 7(10): e47656. DOI:10.1371/journal.pone.0047656 |
| [40] | Weiss S, Xu ZZ, Peddada S, Amir A, Bittinger K, Gonzalez A, Lozupone C, Zaneveld JR, Vázquezbaeza Y, Birmingham A. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome, 2017, 5(1): 27. DOI:10.1186/s40168-017-0237-y |
| [41] | Legendre P. Studying beta diversity: ecological variation partitioning by multiple regression and canonical analysis. Journal of Plant Ecology, 2008, 1(1): 3-8. DOI:10.1093/jpe/rtm001 |
| [42] | Wooley JC, Godzik A, Friedberg I. A primer on metagenomics. PLoS Computational Biology, 2010, 6(2): e1000667. DOI:10.1371/journal.pcbi.1000667 |
| [43] | Namiki T, Hachiya T, Tanaka H, Sakakibara Y. MetaVelvet: an extension of Velvet assembler to de novo metagenome assembly from short sequence reads. Nucleic Acids Research, 2012, 40(20): e155. DOI:10.1093/nar/gks678 |
| [44] | Peng Y, Leung HCM, Yiu SM, Chin FYL. Meta-IDBA: a de novo assembler for metagenomic data. Bioinformatics, 2011, 27(13): i94-i101. DOI:10.1093/bioinformatics/btr216 |
| [45] | Laserson J, Jojic V, Koller D. Genovo: de novo assembly for metagenomes. Journal of Computational Biology, 2011, 18(3): 429-443. DOI:10.1089/cmb.2010.0244 |
| [46] | Vollmers J, Wiegand S, Kaster AK. Comparing and evaluating metagenome assembly tools from a microbiologist's perspective - not only size matters!. PLoS One, 2017, 12(1): e0169662. DOI:10.1371/journal.pone.0169662 |
| [47] | Zhu WH, Lomsadze A, Borodovsky M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Research, 2010, 38(12): e132. DOI:10.1093/nar/gkq275 |
| [48] | Kelley DR, Liu B, Delcher AL, Pop M, Salzberg SL. Gene prediction with Glimmer for metagenomic sequences augmented by classification and clustering. Nucleic Acids Research, 2012, 40(1): e9. DOI:10.1093/nar/gkr1067 |
| [49] | Noguchi H, Park J, Takagi T. MetaGene: prokaryotic gene finding from environmental genome shotgun sequences. Nucleic Acids Research, 2006, 34(19): 5623-5630. DOI:10.1093/nar/gkl723 |
| [50] | Hoff KJ, Lingner T, Meinicke P, Tech M. Orphelia: predicting genes in metagenomic sequencing reads. Nucleic Acids Research, 2009, 37(Web Server issue): W101-W105. |
| [51] | Rho M, Tang HX, Ye YZ. FragGeneScan: predicting genes in short and error-prone reads. Nucleic Acids Research, 2010, 38(20): e191. DOI:10.1093/nar/gkq747 |
| [52] | Liu YC, Guo JT, Hu GQ, Zhu HQ. Gene prediction in metagenomic fragments based on the SVM algorithm. BMC Bioinformatics, 2013, 14(Suppl 5): S12. DOI:10.1186/1471-2105-14-S5-S12 |
| [53] | Trimble WL, Keegan KP, D'Souza M, Wilke A, Wilkening J, Gilbert J, Meyer F. Short-read reading-frame predictors are not created equal: sequence error causes loss of signal. BMC Bioinformatics, 2012, 13: 183. DOI:10.1186/1471-2105-13-183 |
| [54] | Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics, 2006, 22(13): 1658-1659. DOI:10.1093/bioinformatics/btl158 |
| [55] | Qin JJ, Li RQ, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li JH, Xu JM, Li SC, Li DF, Cao JJ, Wang B, Liang HQ, Zheng HS, Xie YL, Tap J, Lepage P, Bertalan M, Batto JM, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P, Sicheritz-Ponten T, Turner K, Zhu HM, Yu C, Li ST, Jian M, Zhou Y, Li YR, Zhang XQ, Li SG, Qin N, Yang HM, Wang J, Brunak S, Doré J, Guarner F, Kristiansen K, Pedersen O, Parkhill J, Weissenbach J, MetaHIT Consortium, Bork P, Ehrlich SD, Wang J. A human gut microbial gene catalogue established by metagenomic sequencing. Nature, 2010, 464(7285): 59-65. DOI:10.1038/nature08821 |
| [56] | The Human Microbiome Project Consortium. A framework for human microbiome research. Nature, 2012, 486(7402): 215-221. DOI:10.1038/nature11209 |
| [57] | Altschul SF, Madden TL, Schäffer AA, Zhang JH, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research, 1997, 25(17): 3389-3402. DOI:10.1093/nar/25.17.3389 |
| [58] | Kent WJ. BLAT——the BLAST-like alignment tool. Genome Research, 2002, 12(4): 656-664. DOI:10.1101/gr.229202 |
| [59] | Wang RH, Mei Y, Xu L, Zhu XW, Wang Y, Guo J, Liu LW. Genome-wide characterization of differentially expressed genes provides insights into regulatory network of heat stress response in radish (Raphanus sativus L.).. Functional & Integrative Genomics, 2018, 18(2): 225-239. |
| [60] | Hinsu AT, Parmar NR, Nathani NM, Pandit RJ, Patel AB, Patel AK, Joshi CG. Functional gene profiling through metaRNAseq approach reveals diet-dependent variation in rumen microbiota of buffalo (Bubalus bubalis). Anaerobe, 2017, 44: 106-116. DOI:10.1016/j.anaerobe.2017.02.021 |
| [61] | Murphy R, Tsai P, Jüllig M, Liu A, Plank L, Booth M. Differential changes in gut microbiota after gastric bypass and sleeve gastrectomy bariatric surgery vary according to diabetes remission. Obesity Surgery, 2017, 27(4): 917-925. DOI:10.1007/s11695-016-2399-2 |
| [62] | Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Research, 2004, 32(Suppl. 1): D277-D280. |
| [63] | Huerta-Cepas J, Szklarczyk D, Forslund K, Cook H, Heller D, Walter MC, Rattei T, Mende DR, Sunagawa S, Kuhn M, Jensen LJ, Von Mering C, Bork P. eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Research, 2016, 44(D1): D286-D293. DOI:10.1093/nar/gkv1248 |
| [64] | The Gene Ontology Consortium. Expansion of the gene ontology knowledgebase and resources. Nucleic Acids Research, 2017, 45(D1): D331-D338. DOI:10.1093/nar/gkw1108 |
| [65] | Liu B, Pop M. ARDB—antibiotic resistance genes database. Nucleic Acids Research, 2009, 37(Suppl. 1): D443-D447. |
| [66] | Lombard V, Ramulu HG, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Research, 2014, 42(D1): D490-D495. DOI:10.1093/nar/gkt1178 |
| [67] | Winnenburg R, Baldwin TK, Urban M, Rawlings C, Köhler J, Hammond-Kosack KE. PHI-base: a new database for pathogen host interactions. Nucleic Acids Research, 2006, 34(Database issue): D459-D464. |
| [68] | Chen LH, Zheng DD, Liu B, Yang J, Jin Q. VFDB 2016: hierarchical and refined dataset for big data analysis--10 years on. Nucleic Acids Research, 2016, 44(D1): D694-D697. DOI:10.1093/nar/gkv1239 |
| [69] | Saier MH Jr, Reddy VS, Tsu BV, Ahmed MS, Li C, Moreno-Hagelsieb G. The Transporter Classification Database (TCDB): recent advances. Nucleic Acids Research, 2016, 44(D1): D372-D379. DOI:10.1093/nar/gkv1103 |
| [70] | Altermann E, Klaenhammer TR. PathwayVoyager: pathway mapping using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. BMC Genomics, 2005, 6: 60. DOI:10.1186/1471-2164-6-60 |
| [71] | Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, Fluckiger A, Messaoudene M, Rauber C, Roberti MP, Fidelle M, Flament C, Poirier-Colame V, Opolon P, Klein C, Iribarren K, Mondragón L, Jacquelot N, Qu B, Ferrere G, Clémenson C, Mezquita L, Masip JR, Naltet C, Brosseau S, Kaderbhai C, Richard C, Rizvi H, Levenez F, Galleron N, Quinquis B, Pons N, Ryffel B, Minard-Colin V, Gonin P, Soria JC, Deutsch E, Loriot Y, Ghiringhelli F, Zalcman G, Goldwasser F, Escudier B, Hellmann MD, Eggermont A, Raoult D, Albiges L, Kroemer G, Zitvogel L. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science, 2018, 359(6371): 91-97. DOI:10.1126/science.aan3706 |
| [72] | Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, Prieto PA, Vicente D, Hoffman K, Wei SC, Cogdill AP, Zhao L, Hudgens CW, Hutchinson DS, Manzo T, Petaccia De Macedo M, Cotechini T, Kumar T, Chen WS, Reddy SM, Szczepaniak Sloane R, Galloway-Pena J, Jiang H, Chen PL, Shpall EJ, Rezvani K, Alousi AM, Chemaly RF, Shelburne S, Vence LM, Okhuysen PC, Jensen VB, Swennes AG, McAllister F, Marcelo Riquelme Sanchez E, Zhang Y, Le Chatelier E, Zitvogel L, Pons N, Austin-Breneman JL, Haydu LE, Burton EM, Gardner JM, Sirmans E, Hu J, Lazar AJ, Tsujikawa T, Diab A, Tawbi H, Glitza IC, Hwu WJ, Patel SP, Woodman SE, Amaria RN, Davies MA, Gershenwald JE, Hwu P, Lee JE, Zhang J, Coussens LM, Cooper ZA, Futreal PA, Daniel CR, Ajami NJ, Petrosino JF, Tetzlaff MT, Sharma P, Allison JP, Jenq RR, Wargo JA. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science, 2018, 359(6371): 97-103. DOI:10.1126/science.aan4236 |