 2017, Vol. 57
2017, Vol. 57中国科学院微生物研究所,中国微生物学会,中国菌物学会
文章信息
- 王斌, 陈庆彩, 胡晓珂. 2017
- Bin Wang, Qingcai Chen, Xiaoke Hu. 2017
- 微生物降解芘过程中的关键细菌
- Key bacteria during microbial degradation of pyrene
- 微生物学报, 57(6): 856-866
- Acta Microbiologica Sinica, 57(6): 856-866
-
文章历史
- 收稿日期:2017-02-16
- 修回日期:2017-04-05
- 网络出版日期:2017-04-05

引用本文 |



2. 中国科学院大学, 北京 100049
2. University of Chinese Academy of Sciences, Beijing 100049, China
根据《中国海洋环境状况公报》(1997–2015),石油类污染物是中国近海海域主要污染物之一[1]。生物降解被认为是污染环境中石油烃降解的重要途径,微生物是最重要的降解者之一[2]。石油组分比较复杂,主要包括饱和烃、芳香烃以及胶质、沥青等非烃类物质。相对于饱和烃,更难被降解的芳香烃具有更高的“三致效应”毒性,属优先检测的有机污染物[3]。四环及四环以上的多环芳烃(polycyclic aromatic hydrocarbons,PAHs)因其结构特殊并具有较高的热力学稳定性而可在环境中持久存在,其“三致效应”更加明显。因此,研究微生物降解多环芳烃的机理机制是当前石油烃生物修复研究领域的重要方向。目前实验室内已驯化分离到数量可观的多环芳烃降解菌,多属于Proteobacteria、Actinobacteria、Cyanobacteria以及Firmicutes[4]。这些微生物在纯培养条件下厌氧降解[5]或好氧降解[6]多环芳烃的机制也已被很好地阐释。值得注意的是,近年的研究认为在复杂微生物群落中广泛存在由不同微生物之间复杂共生关系(互利共生[7]、竞争[8]等)构成的生态网络[9],并且生态网络可以影响微生物群落的变迁[10]。一般认为,存在协同作用的微生物菌群具有相对更高的降解效率[11]。我们的前期研究也表明微生物之间的共生关系是它们共适应降解石油烃及重建稳定微生物生态系统的重要途径[12]。因此,研究石油烃修复过程中微生物群落的变化尤其是微生物之间的互作网络对开发更高效的微生物修复技术具有重要的理论指导意义,同时也有助于更好地理解微生物生态系统如何响应环境污染。
由于存在共生关系的微生物在丰度上表现出一定的相关性,例如互利共生微生物的丰度会表现出正相关[7],而处于竞争关系的微生物的丰度会表现出负相关[8],因此借助微生物的丰度数据我们可以构建不同微生物之间的生态网络进而推测微生物之间的相互作用[9, 13-14]。还可以根据生态网络的拓扑性质了解“关键种”[15]及互作关系。目前关于污染环境中微生物群落结构变化的研究已有很多,但多数仅仅关注了群落组成及相关功能基因的丰度变化。一方面我们认为优势菌是影响群落功能的主要因素,因此研究焦点便会更多地集中在优势菌上。例如在前期研究中,我们通过建立实验室驯化系统研究了芘和苯甲酸钠胁迫后渤海沉积物中微生物群落结构的变化[16]。实验结果表明芘和苯甲酸钠显著改变了沉积物中微生物群落的组成,极大降低了海洋沉积物中菌群的丰度和多样性,其中海杆菌属(Marinobacter)、假海源菌属(Pseudidiomarina)及碳酸噬胞菌属(Aequorivita)得到了极大富集。另一方面,低丰度菌在近几年也被认为对群落功能有巨大贡献。例如Adam等人[17]利用13C标记的芘研究土壤中微生物群落对芘胁迫的响应,结果表明土壤中的降解微生物种类繁多,起到关键作用的并不仅仅是丰度较高的优势菌,还包括数目庞大的低丰度菌。因此如何系统地理解群落功能的执行便成为重要的前沿科学问题,基于系统生物学理论的微生物生态网络是其中一种重要手段:微生物生态网络可用于揭示不同分类的微生物之间的生态互作[9, 14],而网络的拓扑属性被用于研究微生物群落应对环境改变的稳定性[10, 12]。得益于高通量测序技术和生物信息学的迅猛发展,基于16S rDNA的微生物群落数据和各种算法(如eLSA[18],CCLasso[19])极大促进了微生物生态网络研究的发展。同时海量的相关基因组信息及代谢模型数据,也为更深入研究微生物之间的相互作用提供了必要基础[20]。Jeanbille等人[21]通过构建基于Spearman相关性的微生物生态网络研究了法国毗邻地中海及大西洋的近海海域表层沉积物中微生物群落对持续性多环芳烃污染的响应,结果表明环境扰动显著影响了微生物生态网络的结构。然而目前关于多环芳烃降解过程中微生物生态网络的研究仍然不足:已报道的研究总数较少,且其中陆地环境研究较多(如赵慧慧等人的研究[22]表明长期石油污染显著影响了胜利油田周围盐碱化土壤中的微生物分子生态网络的拓扑结构),海洋环境较少。
芘是由4个苯环构成的PAHs,是检测PAHs污染的指示物及研究PAHs生物降解的模型分子[23]。因此本文拟构建室内微宇宙体系,以来自渤海表层沉积物的微生物群落为研究对象,以芘为添加污染物,研究芘降解过程中微生物群落结构变迁,重点研究芘降解过程中的微生物生态网络。本研究将有助于更好地理解功能菌群在生态系统内的相互作用及共适应机制这一前沿科学问题,也将有助于探索如何调节功能菌群使其发挥最大降解潜力。
1 材料和方法 1.1 沉积物采集及处理沉积物于2016年1月17日使用箱式采泥器从渤海(38.95 N 118.95 E)采集,取表层1–2 cm沉积物约500 g置于灭菌烧杯中,于冰盒中搅拌30 min以充分混匀后转移至无菌密封袋中并贮存于4 ℃,48 h内带回实验室进行样品前处理。处理方法如下:取约450 g沉积物再次混匀(方法同上),利用真空冷冻干燥机(岛津,日本)对其中约30 g沉积物进行冷冻干燥,干燥后的样品置于–80 ℃保存用于总DNA提取。
1.2 试剂及培养基二氯甲烷、正己烷及无水硫酸钠均为国药分析纯,甲醇为阿拉丁产分析纯,芘(99.9%)、蒽(99.9%),购自Sigma公司。无机盐培养基(MSM)组成为:(NH4)2SO4 1.0 g,Na2HPO4 0.8 g,KH2PO4 0.2 g,MgSO4·7H2O 0.2 g,FeCl3·3H2O 0.005 g,(NH4)6Mo7O24·4H2O 0.001 g,CaCl2·2H2O 0.1 g,NaCl 20 g,溶于1 L去离子水中,pH为7.2,121 ℃灭菌20 min。芘母液:用二氯甲烷配制芘母液,使其浓度为0.4 g/mL。蒽母液:二氯甲烷配制,浓度为0.01 g/mL。
1.3 微宇宙实验设计实验设计见表 1。取63个250 mL锥形瓶(已灭菌),设对照组(21个)和实验组(42个)。向各瓶中加入100 mL灭菌MSM培养基、100 µL芘母液,使芘的终浓度为0.4 g/L,待二氯甲烷完全挥发后使用。向实验组各瓶加入10 g沉积物,并将对照组及实验组所有瓶置于摇床中培养(30 ℃,60 r/min)。分别在第0、3、6、12、18、24、30天取样,每次取3个对照组,6个实验组。其中3个对照组及3个实验组用于萃取分析芘降解率,3个实验组冷冻干燥后保存于–80 ℃,用于总DNA提取。
| Sampling time/d | Samples | Purpose | |
| Control | 0, 3, 6, 12, 18, 24, 30 | 3 | Chemical analysis |
| Treatments | 0, 3, 6, 12, 18, 24, 30 | 6 | 3 for chemical analysis and 3 for DNA extraction |
1.4 芘降解率测定
以蒽为内标参照我们的前期研究[12]进行降解率的测定。主要包括超声萃取后过滤收集液相、分液漏斗收集有机相并用无水硫酸钠除水、旋蒸后用甲醇进行溶剂置换并定容至15 mL、0.45 µm滤膜过滤并–20 ℃保存、HPLC检测。检测条件为:流动相(乙腈:水)=80:20;流速1.0 mL/min;温度(T) 30 ℃;检测波长254 nm;进样量10 µL;色谱柱为C18反相柱(4.6 mm×250 mm,SunFire)。降解率计算公式为:
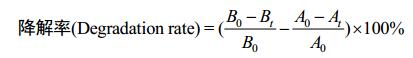
|
A:对照组中芘的含量(A0,0天芘的含量;At,t天芘的含量);
B:实验组中芘的含量(B0,0天芘的含量;Bt,t天芘的含量)。
1.5 DNA提取及测序DNA提取使用MoBio PowerSoilTM kit (MoBio,Carlsbad,CA,USA)试剂盒并参照使用说明进行。本研究共7个取样时间(T0 d、T3 d、T6 d、T12 d、T18 d、T24 d、T30 d),每个时间点3个平行,共计21个DNA样品。样品被送至北京诺禾致源公司(北京,中国),使用Illumina Hiseq 2500对V4-V5高变区进行测序。原始序列已上传至SRA数据库,样品编号为SAMN06216651– SAMN06216671。
1.6 生物信息学分析使用USEARCH 9.0[24]对原始序列进行聚类分析,主要步骤包括双末端序列装配、去除低质量序列、去重复、去嵌合体、在97%相似性水平上进行操纵分类单元(operational taxonomic unit,OTU)聚类分析、全局比对后生成含有OTU在各样品中序列数的表格,以上步骤的参数采用默认推荐参数值。使用Greengenes (version 13.8) 数据库对获得的OTUs代表序列进行分类水平的鉴定。由于引物是面向细菌的V4-V5区进行设计,且只有6个OTUs被鉴定为古菌(远低于古菌的实际种类数),因此在界水平上被分类为古菌及不能被分类的OTUs将被移除。利用Mothur软件[25]使用重抽样的方式将所有样品的测序深度标准化到49000条序列并计算Good’s coverage、Shannon-Wiener、Chao1指数,运算执行1000次后取平均值。计算各OTUs在各样品中的相对丰度并分别在纲及属水平上进行汇总。基于非参数Kruskal-Wallis秩和检验统计在T0 d和T30 d中相对丰度存在显著差异的OTUs (P<0.05),将这些OTUs称为差异OTUs。根据它们的相对丰度变化情况,将差异OTUs分为升高组(increased OTUs,i-OTUs)与降低组(decreased OTUs,d-OTUs),汇总T0 d及T30 d的i-OTUs及d-OTUs数目及总相对丰度,并利用气泡图可视化二者在分类水平上的汇总信息。鉴于我们的研究对象为可能影响芘生物降解的细菌,因此只有在T30 d的样品中平均相对丰度大于0.05%的OTUs被纳入分析。基于OTUs的相对丰度及CCLasso算法[19]计算微生物之间的相关性(r>0,P<0.05) 并构建微生物生态网络(Microbial ecological network,MEnet);统计在MEnet中i-OTUs与d-OTUs的数目。上述分析中如无特别指出,计算及绘图均借助R软件[26]进行。
对未能在属水平上获得分类信息的OTUs (属未知OTUs),借助本地BLAST 2.2.22程序、OTUs的代表序列及ModelSeed数据库(http://modelseed.org/)获得近源基因组(16S序列相似性>97%)的物种信息并以此补充属未知OTUs的属水平分类信息[27]。构建属水平MEnet (g-MEnet),则网络中的点代表属,边代表连接的2个属之间存在相关的OTUs的对数(定义为NC值)。根据网络的拓扑属性探究重要菌群及他们之间的关系。
2 结果和分析 2.1 芘的降解率变化趋势对照组的芘含量在30 d内损失不足1%,表明芘的降解主要由微生物降解驱动。如图 1-A所示,实验组芘的降解率在30 d内持续增加且在30 d时达到(67.07±2.37)%,但降解速率(即降解率的斜率)以3 d为界呈现先增加后降低的趋势。

|
| 图 1. 芘降解率(A)、OTUs获得数(B)、Chao1(C)及Shannon-Wiener(D)随时间变化趋势 Figure 1. The line chart illustrated the tendency of degradation rate (A), number of observed OTUs (B), Chao1 indices (C) and Shannon-Wiener indices (D) over time. |
2.2 微生物群落结构及变迁
经过质控及筛选,21个样品共获得1330269条序列;在97%相似性水平上共获得3712个OTUs。在所有样品中,代表覆盖度水平的Good’s coverage指数均大于98%,表明本研究的测序深度可以很好地反映微生物群落的多样性及群落结构。代表群落物种丰度水平的OTUs获得数(图 1-B)及Chao1指数(图 1-C)在0 d分别为2475.12±94.18、3170.59±67.53,而在30 d则为972.89±180.39、1547.04±217.84,总体趋势表现为持续降低。可同时反映多样性及均匀度的Shannon-Wiener指数(图 1-D)在0 d的值为5.52±0.21,在12 d达到最低值2.98±0.87,随后升高并于第24天趋于稳定(24 d:3.95±0.16,30 d:3.83±0.34,P>0.05)。
在纲水平上,沉积物中的优势菌(0 d)主要为Alphaproteobacteria [(8.86 ± 1.57) %]、Deltaproteobacteria [(24.72±4.12) %]以及Gammaproteobacteria [(36.73±6.10)%]。30 d后,优势菌主要为Alphaproteobacteria [(45.05±14.83)%]、Gammaproteobacteria [(30.68±7.04)%]及Flavobacteriia [(7.25±6.62)%] (图 2-A)。在属的水平上,沉积物中的优势菌主要包括Desulfococcus [(5.16 ± 1.03) %]、Pseudidiomarina [(2.63±0.79)%]及Marinobacter [(2.54±0.91)%]等,经过30 d培养后的优势属为Flavobacterium [(4.84 ±1.91) %]、Bacillus [(4.46±1.83)%]及Marinobacter [(3.45±1.28)%]等(图 2-B)。通过统计i-OTUs及d-OTUs的分类信息表明:(1) 共计102个i-OTUs与1551个d-OTUs,与前文alpha多样性降低的结果一致;(2) 对i-OTUs而言,在纲水平上主要属于Alphaproteobacteria、Flavobacteriia、Planctomycetia及Bacilli (图 2-C),在属水平上主要属于Flavobacterium、Bacillus、Planctomyces及Erythrobacter等(图 2-D);(3) 对d-OTUs而言,在纲水平上主要属于Deltaproteobacteria、Gammaproteobacteria、Anaerolineae及Spirochaetes等(图 2-E),在属水平上主要属于Desulfococcus、Spirochaeta及Pseudoalteromonas等(图 2-F)。

|
| 图 2. 微生物群落结构组成及差异OTUs的分类信息汇总 Figure 2. The taxonomic summary of bacterial compositions and the differential OTUs. A: the bacterial compositions of samples at class level, those classes with relative abundances less than 1% in 0 d were grouped into "Others"; B: the bacterial compositions of samples at genus level, only the genera with relative abundances more than 0.5% in either 0 d or 30 d were shown here; C–F: the bubble diagram revealed the relative abundances and numbers of the differential OTUs affiliated to different classes/genera in 0 d and 30 d: i-OTUs at class level (C); i-OTUs at genus level (D); d-OTUs at class level (E); d-OTUs at genus level (F). |
2.3 微生物生态网络分析
筛选后共计127个OTUs被用于预测MEnet,结果表明MEnet由53个点及317条边组成,其中属于i-OTUs与d-OTUs的分别为37个与0个(表 2)。在g-MEnet中,共有29个点143条边(图 3)。点度最高的点为Unclassified Flavobacteriaceae (degree=27),其次为Unclassified alpha proteobacterium Q-1 (degree=19) 及Planctomyces (degree=19) 等(表 3)。NC值最高的边为Erythrobacter-Unclassified Flavobacteriaceae (NC=17),其次为Erythrobacter-Unclassified Alphaproteobacteria (NC=15)、Erythrobacter-Unclassified alpha proteobacterium Q-1 (NC=12) 等(表 3)。
| Attributes | Number | i-OTUs | d-OTUs |
| Nodes | 53 | 37 | 0 |
| Edges | 317 | NA | NA |

|
| 图 3. 芘降解过程中的属水平微生物生态网络g-MEnet Figure 3. The network g-MEnet revealed the associations among selected OTUs at genus level. The size of the circles and the width of edges were positively correlated with the degree values and the NC values, respectively. |
| Attributes | Informations |
| Nodes | 29 |
| Nodes (degree > 15) | Unclassified Flavobacteriaceae: 27 Unclassified alpha proteobacterium Q-l: 19 Planctomyces: 19 Unclassified Alphaproteobacteria: 16 Sneathiella: 16 Erythrobacter: 15 |
| Edges | 143 |
| Edges (NC > 5) | Erythrobacter-Unclassified Flavobacteriaceae: 17 Erythrobacter-Unclassified Alphaproteobacteria: 15 Erythrobacter-Unclassified alpha proteobacterium Q-l: 12 Erythrobacter-Unc: 10 Unclassified alpha proteobacterium Q-l-Unclassified Alphaproteobacteria: 9 Erythrobacter-Erythrobacter. 8 Unclassified alpha proteobacterium Q-l-Unclassified Flavobacteriaceae: 8 Unclassified Flavobacteriaceae-Unclassified Alphaproteobacteria: 7 Erythrobacter-Maricaulis: 6 Erythrobacter-Muricauda: 6 Unc-Unclassified Alphaproteobacteria: 6 Erythrobacter-Planctomyces: 5 Erythrobacter-Unclassificd Flavobacteriia: 5 Erythrobacter-Unclassified Solibacteres: 5 Unclassified Flavobacteriaceae-Unclassified Flavobacteriaceae: 5 |
3 讨论
一般来说,包括芘在内的石油烃对微生物群落的影响主要体现在两个方面:一是它们可以作为部分细菌生长所需的唯一碳源,如Bacillus、Cyanobacteria、Cycloclasticus、Flavobacterium、Marinobacter、Mycobacterium、Planctomyces、Pseudomonas、Pseudoalteromonas、Pseudidiomarina、Rhodococcus和Vibrio等[28];另一方面,它们对某些微生物具有毒性。因此在受到石油烃胁迫后,微生物群落结构将发生明显改变[12]。我们前期的研究表明,添加芘胁迫后,渤海表层沉积物中的微生物群落结构发生了明显改变[12, 16]:一部分可能具有降解功能的微生物的相对丰度明显增加,另一部分微生物的相对丰度明显降低甚至变为0进而引起相关群落生态功能的缺失。因此通过分析微生物相对丰度的变化趋势,我们可以推测芘降解过程中的潜在功能菌,也可以获知胁迫对群落生态功能的影响。本研究中,属于i-OTUs的Flavobacterium、Bacillus、Planctomyces等的多环芳烃降解能力均已有报道[28]。值得注意的是,58.8%的i-OTUs属于Alphaproteobacteria,但它们在属水平上的分类信息多为未知,提示我们对渤海沉积物中多环芳烃降解菌的认识仍非常缺乏。沉积物中的Deltaproteobacteria (如本研究中明显降低的Desulfococcus属)多参与硫酸盐还原[29],同时在沉积物环境中,石油烃的厌氧降解多与硫循环过程耦合[30]。因此本研究中Deltaproteobacteria的相对丰度显著降低,一方面可能与Deltaproteobacteria多厌氧而本研究的培养条件存在氧气输入有关,另外一方面也暗示多环芳烃污染对渤海沉积物中的硫循环过程存在一定的不利影响。
由于绝大多数微生物的不可培养性,使得我们无法采用纯培养的方式获取微生物之间的共生关系。但由于共生关系在丰度相关性上的体现,使得我们可以基于微生物群落组成及它们的相对丰度变化趋势预测微生物之间的共生关系[9–10, 14–15]。微生物之间的共生关系表现在丰度相关性上可以是正相关也可以是负相关。在环境中,非生物因素(如营养盐水平)的生境选择和生物因素(微生物之间的相互作用)是影响丰度水平的重要因素[10, 31–32]。因此,不同细菌之间存在丰度正相关可能是由于他们之间存在功能互补如互利共生(直接相关),也可能是由于它们有相似的环境要求(间接相关,如资源丰富的有利环境引起的丰度同步增加或者不利环境引起的丰度同步降低均可在相关性上表现出正相关)。值得注意的是,传统计算相关性的算法(如Pearson相关,Spearman相关等)并不能区分直接相关还是间接相关。这样不仅会增加分析的复杂性,还会在分析生态网络的拓扑属性时带来更大的误差。本研究使用的CCLasso算法[19]借助最小二乘法和增广拉格朗日乘子法移除第三方效应,使得构建的生态网络不包含间接相关。在MEnet中不存在属于d-OTUs的点,表明由于芘胁迫引起的相对丰度同步降低而表现出正相关的OTUs并未包含在我们构建的生态网络中。因此本研究中在丰度变化上表现出正相关的微生物之间可认为存在互利共生或者协同作用。在T30 d时平均相对丰度大于0.05%的i-OTUs共计49个,而在MEnet中属于i-OTUs的共有37个,表明大多数的i-OTUs的相对丰度变化不仅和芘有关,还和它们之间可能存在的互利共生或协同作用有关。
寻找“关键种”是微生物生态领域的关键科学问题,基于微生物生态网络的拓扑属性(主要指点度中心度:在1个网络中与1个点直接相连的其他点的个数)寻找核心微生物是目前比较认可的方法[9, 14–15]。基于点度中心度,本研究样品中的核心微生物包括Planctomyces, Erythrobacter等(表 3)。此外,代表互作关系的边是研究生态网络的另一个重要内容[33]。为了评价2个不同属微生物之间的互作强度,我们引入NC值进行评价。NC值受以下2个方面因素的影响:每个属G所含OTUs的数目N;2个不同属微生物之间存在互作的倾向性P。N和P的值越高,NC值越高。一方面N值越高意味着属G在T30 d时所占比例越高,则属G在芘降解过程中越趋向于重要;另一方面P值越高两个属之间越倾向于互作。因此在生态网络中NC值越高的边越趋向于重要。值得注意的是,在g-MEnet中NC值较高的边(NC>5) 多连接到属Erythrobacter。综合点度中心度我们认为Erythrobacter为本研究中最重要的微生物类群,在我们的前期研究中发现,Erythrobacteraceae的微生物是重要的多环芳烃降解者[12]。
| [1] | State Oceanic Administration. China Oceanic Information Network. 国家海洋局. 中国海洋信息网. http://www.coi.gov.cn/gongbao/huanjing/. [2017-02-10]. |
| [2] | Head IM, Jones DM, Röling WFM. Marine microorganisms make a meal of oil. Nature Reviews Microbiology, 2006, 4(3): 173–182 DOI:10.1038/nrmicro1348. |
| [3] |
Huang GP, Chen YJ, Lin T, Tang JH, Liu DY, Li J, Zhang G. The distribution and ecological risk of polycyclic aromatic hydrocarbons of surface sediments in the intertidal zone of Bohai Bay, China.
China Environmental Science, 2011, 31(11): 1856–1863 (in Chinese) 黄国培, 陈颖军, 林田, 唐建辉, 刘东艳, 李军, 张干. 渤海湾潮间带表层沉积物中多环芳烃的含量分布和生态风险. 中国环境科学, 2011, 31(11): 1856–1863. |
| [4] | Louvado A, Gomes NC, Simões MMQ, Almeida A, Cleary DFR, Cunha A. Polycyclic aromatic hydrocarbons in deep sea sediments:microbe-pollutant interactions in a remote environment. Science of the Total Environment, 2015, 526: 312–328 DOI:10.1016/j.scitotenv.2015.04.048. |
| [5] |
Sun MM, Teng Y, Luo YM. Progresses in anaerobic biodegradation of polycyclic aromatic hydrocarbons-a review.
Acta Microbiologica Sinica, 2012, 52(8): 931–939 (in Chinese) 孙明明, 滕应, 骆永明. 厌氧微生物降解多环芳烃研究进展. 微生物学报, 2012, 52(8): 931–939. |
| [6] | Baboshin MA, Golovleva LA. Aerobic bacterial degradation of polycyclic aromatic hydrocarbons (PAHs) and its kinetic aspects. Microbiology, 2012, 81(6): 639–650 DOI:10.1134/S0026261712060021. |
| [7] | Molloy S. Environmental microbiology:disentangling syntrophy. Nature Reviews Microbiology, 2014, 12(1): 7 |
| [8] | Hibbing ME, Fuqua C, Parsek MR, Peterson SB. Bacterial competition:surviving and thriving in the microbial jungle. Nature Reviews Microbiology, 2010, 8(1): 15–25 DOI:10.1038/nrmicro2259. |
| [9] | Faust K, Raes J. Microbial interactions:from networks to models. Nature Reviews Microbiology, 2012, 10(8): 538–550 DOI:10.1038/nrmicro2832. |
| [10] | Gokul JK, Hodson AJ, Saetnan ER, Irvine-Fynn TDL, Westall PJ, Detheridge AP, Takeuchi N, Bussell J, Mur LAJ, Edwards A. Taxon interactions control the distributions of cryoconite bacteria colonizing a High Arctic ice cap. Molecular Ecology, 2016, 25(15): 3752–3767 DOI:10.1111/mec.2016.25.issue-15. |
| [11] | Wang FK, Li C, Wang HJ, Chen WL, Huang QY. Characterization of a phenanthrene-degrading microbial consortium enriched from petrochemical contaminated environment. International Biodeterioration & Biodegradation, 2016, 115: 286–292 |
| [12] | Wang H, Wang B, Dong WW, Hu XK. Co-acclimation of bacterial communities under stresses of hydrocarbons with different structures. Scientific Reports, 2016, 6: 34588 DOI:10.1038/srep34588. |
| [13] | Barberán A, Bates ST, Casamayor EO, Fierer N. Using network analysis to explore co-occurrence patterns in soil microbial communities. The ISME Journal, 2012, 6(2): 343–351 DOI:10.1038/ismej.2011.119. |
| [14] | Fuhrman JA, Cram JA, Needham DM. Marine microbial community dynamics and their ecological interpretation. Nature Reviews Microbiology, 2015, 13(3): 133–146 DOI:10.1038/nrmicro3417. |
| [15] | Zhou JZ, Deng Y, Luo F, He ZL, Yang YF. Phylogenetic molecular ecological network of soil microbial communities in response to elevated CO2. mBio, 2011, 2(4): e00122–11 |
| [16] |
Sun XN, Hu XK, Wang H. Response of bacterial communities to pyrene and benzoate sodium in marine sediments.
Oceanologia et Limnologia Sinica, 2015, 46(6): 1304–1311 (in Chinese) 孙向楠, 胡晓珂, 王慧. 海洋沉积物细菌群落结构对芘和苯甲酸钠胁迫的响应. 海洋与湖沼, 2015, 46(6): 1304–1311. |
| [17] | Adam IK, Duarte M, Pathmanathan J, Miltner A, Brüls T, Kästner M. Microbial communities in pyrene amended soil-compost mixture and fertilized soil. AMB Express, 2017, 7(1): 7 DOI:10.1186/s13568-016-0306-9. |
| [18] | Xia LC, Steele JA, Cram JA, Cardon ZG, Simmons SL, Vallino JJ, Fuhrman JA, Sun FZ. Extended local similarity analysis (eLSA) of microbial community and other time series data with replicates. BMC Systems Biology, 2011, 5(S2): S15 |
| [19] | Fang HY, Huang CC, Zhao HY, Deng MH. CCLasso:correlation inference for compositional data through Lasso. Bioinformatics, 2015, 31(19): 3172–3180 DOI:10.1093/bioinformatics/btv349. |
| [20] | Mendes-Soares H, Mundy M, Soares LM, Chia N. MMinte:an application for predicting metabolic interactions among the microbial species in a community. BMC Bioinformatics, 2016, 17: 343 DOI:10.1186/s12859-016-1230-3. |
| [21] | Jeanbille M, Gury J, Duran R, Tronczynski J, Agogué H, Saïd O, Ghiglione JF, Auguet JC. Response of core microbial consortia to chronic hydrocarbon contaminations in coastal sediment habitats. Frontiers in Microbiology, 2016, 7: 1637 |
| [22] |
Zhao HH, Xiao X, Pei M, Zhao Y, Liang YT. Effect of long-term oil contamination on the microbial molecular ecological networks in saline-alkali soils.
Environmental Science, 2016, 37(9): 3582–3589 (in Chinese) 赵慧慧, 肖娴, 裴孟, 赵远, 梁玉婷. 长期石油污染对盐碱化土壤中微生物群落分子生态网络的影响. 环境科学, 2016, 37(9): 3582–3589. |
| [23] | Haritash AK, Kaushik CP. Biodegradation aspects of polycyclic aromatic hydrocarbons (PAHs):a review. Journal of Hazardous Materials, 2009, 169(1/3): 1–15 |
| [24] | Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics, 2010, 26(19): 2460–2461 DOI:10.1093/bioinformatics/btq461. |
| [25] | Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Applied and Environmental Microbiology, 2013, 79(17): 5112–5120 DOI:10.1128/AEM.01043-13. |
| [26] | R Core Team. R:The R Project for Statistical Computing. Vienna, Austria:R Foundation for Statistical Computing, 2016. https://www.R-project.org/. |
| [27] | Konstantinidis KT, Tiedje JM. Prokaryotic taxonomy and phylogeny in the genomic era:advancements and challenges ahead. Current Opinion in Microbiology, 2007, 10(5): 504–509 DOI:10.1016/j.mib.2007.08.006. |
| [28] |
Guo CL, Zheng TL, Hong HS. Biodegradation and bioremediation of polycyclic aromatic hydrocarbons.
Marine Environmental Science, 2000, 19(3): 24–29 (in Chinese) 郭楚玲, 郑天凌, 洪华生. 多环芳烃的微生物降解与生物修复. 海洋环境科学, 2000, 19(3): 24–29. |
| [29] | Liu JW, Liu XS, Wang M, Qiao YL, Zheng YF, Zhang XH. Bacterial and archaeal communities in sediments of the north Chinese marginal seas. Microbial Ecology, 2015, 70(1): 105–117 DOI:10.1007/s00248-014-0553-8. |
| [30] | Moretto LM, Silvestrib S, Ugo P, Zorzi G, Abbondanzi F, Baiocchi C, Iacondini A. Polycyclic aromatic hydrocarbons degradation by composting in a soot-contaminated alkaline soil. Journal of Hazardous Materials, 2005, 126(1/3): 141–148 |
| [31] | Steele JA, Countway PD, Xia L, Vigil PD, Beman JM, Kim DY, Chow CET, Sachdeva R, Jones AC, Schwalbach MS, Rose JM, Hewson I, Patel A, Sun FZ, Caron DA, Fuhrman JA. Marine bacterial, archaeal and protistan association networks reveal ecological linkages. The ISME Journal, 2011, 5(9): 1414–1425 DOI:10.1038/ismej.2011.24. |
| [32] | Nemergut DR, Schmidt SK, Fukami T, O'Neill SP, Bilinski TM, Stanish LF, Knelman JE, Darcy JL, Lynch RC, Wickey P, Ferrenberg S. Patterns and processes of microbial community assembly. Microbiology and Molecular Biology Reviews, 2013, 77(3): 342–356 DOI:10.1128/MMBR.00051-12. |
| [33] | Vályi K, Mardhiah U, Rillig MC, Hempel S. Community assembly and coexistence in communities of Arbuscular mycorrhizal fungi. The ISME Journal, 2016, 10(10): 2341–2351 DOI:10.1038/ismej.2016.46. |